
STXBP1. This is the Epilepsiome page for STXBP1, one of the most common genetic etiologies for epilepsy and neurodevelopmental disorders. While STXBP1 was implicated in human disease in individuals with Ohtahara Syndrome in 2008, the phenotypic spectrum has greatly expanded and is recognized as a prominent gene for epileptic encephalopathies with clinical features including but not limited to neonatal seizures, infantile spasms, focal epilepsy, movement disorders, and neurodevelopmental differences with or without epilepsy.
Here are the most recent blog posts that mention STXBP1
- STXBP1-related disorders: deciphering the phenotypic code
- A disease concept model for STXBP1-related disorders
- Unlocking STXBP1 through Electronic Medical Records
- The 2022 STXBP1 Summit – a personal reflection
- Understanding development and seizures in STXBP1 disorders
In a nutshell. Disease-causing variants in STXBP1 are among the most common causes of epilepsy and neurodevelopmental disorders, with an estimated incidence of approximately 1:30,000. Historically, the phenotypic complexity and heterogeneity of STXBP1-related disorders have limited understanding of clear genotype-phenotype correlations. Furthermore, the association of clinical features with the underlying disease mechanism is less clear than in some other disorders such as ion-channel related disorders. However, in the past few years, an increased focus on this gene, dedicated efforts with an expanded scope of clinical studies, and developments in gene therapies underway, STXBP1 has become one of the neurodevelopmental genes with the “fastest growing knowledge.”
Phenotypes | Genetics | Mechanism & function | Community & resources
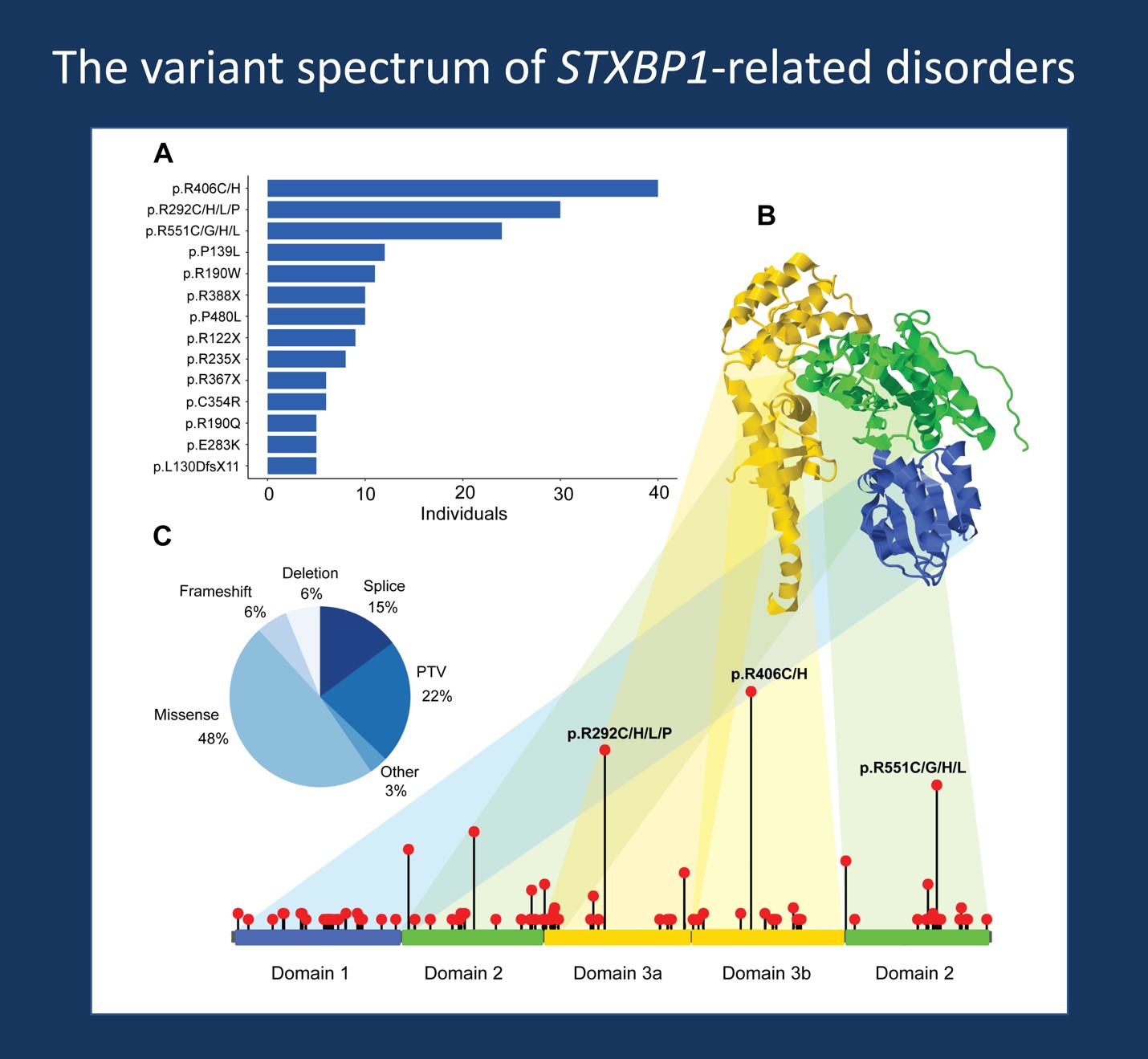
Figure 1. Spectrum of disease-causing variants in STXBP1, highlighting the distribution of recurrent variants found in 534 individuals with STXBP1-related disorders (Xian et al, 2021) across all domains in the gene and overview of variant types including missense variants and protein-truncating variants and deletions in STXBP1.
Phenotypes
General overview. The range of phenotypic presentations associated with STXBP1-related disorders is perplexing. In 2008, STXBP1 was initially discovered and associated exclusively with Ohtahara Syndrome, a severe neonatal epilepsy. However, the phenotypic spectrum has significantly expanded since the initial description by Saitsu and collaborators. In 2015, STXBP1 became recognized as one of the most common genetic etiologies for epileptic encephalopathy. The emergence of case-cohort studies reporting novel associations with numerous disease entities challenged understanding of the full picture of STXBP1-related disorders. While the full range of neurodevelopmental conditions may still not be fully recognized, here is an overview of our current understanding of broad phenotypic subgroups when assessing phenotypes of 534 individuals with STXBP1-related disorders:
Ohtahara Syndrome. The severe presentation of early infantile epileptic encephalopathy (EIEE) with tonic seizures and burst suppression EEG, now known as Ohtahara Syndrome (OS), was the first STXBP1 phenotype to be described. However, as of 2023, OS is now recognized to constitute a smaller subgroup, reported in 49 (9.2%) in our study of 534 individuals with STXBP1-related disorders. Because 90% of our cohort consisted of individuals with epilepsy, it is likely to that the estimate of OS is even lower in frequency in the overall STXBP1 population. Of individuals with OS, the variant spectrum is roughly split with 51% of individuals having missense variants in STXBP1 and 48% having protein-truncating variants (PTV) or deletions. 12% of individuals with the recurrent variant p.Arg406Cys/His were noted to have OS. Other genetic etiologies for OS include ARX, CDKL5, and SLC25A22; however, other causes also include brain abnormalities, metabolic disorders, and unknown causes.
West Syndrome. While a subset of individuals OS later present with a phenotype consistent with West Syndrome, this subgroup also includes individuals with infantile spasms as the first seizure presentation of STXBP1-related disorders. Infantile spasms are among the most common seizure types in STXBP1-related disorders, and the onset risk of developing infantile spasms is recognized to be higher in individuals with PTV and deletions in STXBP1. Preliminary data estimate that up to a half of individuals with neonatal seizures or early infantile seizures develop epileptic spasms. The median age of onset of spasms is 4 months of life, with 90% cumulative onset by 6 months. Of the 77 (14.4%) individuals with West Syndrome and spasms as the first seizure type, 59.7% had PTV/deletions while 39% had missense variants.
Early-Onset Epileptic Encephalopathy (EOEE). The broad phenotypic subgroup of EOEE constituted the largest subgroup within STXBP1-related disorders, assigned to 163 of the 534 individuals included in our study (30.5%). While there may be phenotypic overlap, we refer to EOEE for the subset of individuals with seizure onset in the first three months of life and features of developmental and epileptic encephalopathy (DEE) without a clinical presentation consistent with the pattern of OS or West Syndrome. This cohort represents a more historical label associated with STXBP1-related epilepsy and spans diagnostic groupings. For example, this includes individuals with Early Myoclonic Epilepsy (EME), in addition to a wide range of associated clinical features such as developmental delay, and/or spasticity and movement disorders.
Other Epilepsies. It is estimated that 70% to 80% of individuals with STXBP1-related disorders have epilepsy, however given the range of epilepsy presentations, we refer to this broad subgroup for individuals with other epilepsies that does not fit the pattern of any of the above clinical entities (OS, West Syndrome, or EOEE). Accordingly, this group includes individuals with DEE with seizure onset after the first three months of life. For example, disease causing variants in STXBP1 have been found in individuals with Dravet Syndrome, however these subsets represent less frequent phenotypes. Overall, seizures are prominent in STXBP1-related disorders in the first year of life, with 90% onset by 14 months. However, epilepsy progression is dynamic and presents heterogeneously during this time, in contrast to other genetic epilepsies such as Dravet Syndrome. The most frequent seizure types associated with STXBP1-related disorders are focal-onset seizures and epileptic spasms. In contrast to other genetic epilepsies, several seizure presentations including absence seizures, hemiclonic seizures, and status epilepticus have relatively low frequencies in STXBP1-related disorders.
Neurodevelopmental Disorders. In 2023, individuals with developmental differences without epileptic encephalopathies is increasingly recognized as constituting a larger proportion of individuals with STXBP1-related disorders. This cohort includes individuals with developmental delay, autism, or intellectual disability without epilepsy or with seizures that did not result in a predominant epilepsy phenotype. For example, we would include an individual with focal epilepsy that is controlled by anti-seizure medications and learning differences in this subgroup. 129 (24.2%) individuals were included in this subgroup in our study of 534 individuals with STXBP1-related disorders.
Other phenotypes. The majority of individuals with STXBP1-related disorders have hypotonia, developmental delay, and challenges in motor and language development. A proportion of individuals has been noted to have non-epileptic movement disorders, including truncal and limb ataxia, generalized tremors, and dystonia. Furthermore, a small subset of individuals has been associated with an atypical Rett-like presentation. It has also been suggested that a proportion of individuals with STXBP1 without epilepsy remain undiagnosed, including a fraction of individuals with cerebral palsy. Of note, other non-neurological features including gastrointestinal and respiratory symptoms that were previously underrepresented in the literature have been identified through a disease concept model to impact the lived experience and daily lives of many families with STXBP1.
Summary. Given the phenotypic heterogeneity of STXBP1-related disorders, defining distinct clinical subgroups is not straightforward. There is substantial overlap between subgroups and no single phenotypic feature that can clearly distinguish between disease entities. Nevertheless, when analyzing the larger population of 534 individuals with STXBP1-related disorders, we can provide broad strokes that capture groups of individuals that share higher phenotypic resemblance than expected by chance when compared to the remainder of the cohort. These subgroups include: Ohtahara Syndrome (OS), West Syndrome, EOEE, other epilepsy and DEEs, and neurodevelopmental disorders.
Genetics
Gene-disease validity. There is very strong supporting evidence for the role of STXBP1 in human epilepsy. Under the current gold standard framework for gene curation by the Clinical Genome (ClinGen) consortium, the association of STXBP1 in epilepsy is considered to be “Definitive,” indicating that sufficient evidence has been accumulated with replication over time to support an association with the disease phenotype, considering genetic evidence in addition to experimental evidence. For STXBP1, gene-disease validity is established as a bona fide etiology for human epilepsy.
Variant spectrum. Over two hundred unique disease-causing variants have been identified so far in individuals with STXBP1-related disorders. These variants are distributed across the STXBP1 gene and represent a roughly 50-50 split between individuals with PTV or deletions (including splice site, frameshift, nonsense variants) and individuals with missense variants in STXBP1. More than 50 recurrent variants have been identified, with p.Arg406His/Cys, p.Arg292Cys/His/Leu/Pro, and p.Arg551Cys/Gly/His/Leu as the most common recurrent variants. Recurrent variants identified in five or more individuals are currently known to account for up to a third of all reported individuals with STXBP1-related disorders. Most variants in STXBP1 are found to be de novo. The penetrance of STXBP1-associated phenotypes is high, however individuals with STXBP1-related disorders can have a wide range of phenotypic features and fall along a wide spectrum of disease severity. While most PTV and deletions lead to haploinsufficiency, or loss of a functional copy of the gene, the functional consequence of many missense variants is not clear from the variant itself (see below for disease mechanisms).
Genotype-phenotype correlation. Historically, the range of phenotypic features and baseline genetic heterogeneity have challenged recognition of genotype-phenotype associations. However, there is now emerging evidence that support nominally significant correlations between specific phenotypic features and variant subgroups. It is recognized that individuals with PTV or deletions in STXBP1 have up to a 2.5-fold increased risk for infantile spasms and West Syndrome. In contrast, there is an increased for spastic tetraplegia and burst-suppression EEG in individuals with the recurrent p.Arg406His/Cis missense variants. A nominal increase in risk of focal impaired awareness seizures has also been seen in individuals with p.Arg292/Cys/His/Leu/Pro variants. In contrast, individuals with p.Arg551Cys/Gly/His/Leu variants have an increased risk of generalized tonic-clonic seizures and movement disorders. However, these associations are not black-and-white; high clinical variability is observed even between individuals with the same variant, and none of the recurrent variants showed significant clinical resemblance despite emerging phenotypic signatures.
Mechanism & Function
Protein function. STXBP1 encodes syntaxin-binding protein 1, also known as MUNC18-1, which is critical to synaptic vesicle exocytosis in the presynapse and enables vesicles to fuse with the plasma membrane, facilitating communication between neurons. As a key regulatory protein, MUNC18 orchestrates the assembly of core components of the SNARE (soluble NSF attachment protein receptor) complex. Accordingly, STXBP1-related disorders fall under the umbrella term of SNAREopathies, referring to a group of neurodevelopmental conditions caused by variants in genes encoding components and regulators of the SNARE complex.
Disease mechanisms. The main disease mechanism is haploinsufficiency resulting from heterozygous variants disrupting one functional copy of the STXBP1 gene. However, more recently, dominant-negative effects have been suggested as an alternate mechanism of disease for several missense variants. When assessing a selection of recurrent variants, including p.Arg406His, Guiberson and collaborators find a functional depletion of STXBP1 that is lower than what would be expected by haploinsufficiency alone. Taken together, while haploinsufficiency remains the predominant disease mechanism, further resolution of variant functional consequences will remain critical and provide necessary information to inform the development of targeted therapies.
Preclinical models. To investigate the underlying pathophysiology, preclinical model systems for STXBP1 and research focusing on delineating the underlying mechanism range from haploinsufficiency mouse and macque models to analyze the neuronal activity and relationship between synaptic excitation and inhibition in the brain, to patient-derived iPSC cell lines to understand differences in transcriptomic and gene networks. When comparing and assessing preclinical models, it is important to evaluate the degree to which the studied model system recapitulates aspects of the disease phenotype and whether the genetic mechanism and pathophysiology resulting in a phenotype is consistent. Nevertheless, integration of knowledge from a range of model systems will provide more comprehensive insight into the underlying disease mechanisms and refine identification of molecular targets for precision medicine approaches.
Community & Resources
Family advocacy organizations. The STXBP1 Disorders Foundation is a non-profit organization for STXBP1 families and researchers. They have various resources and support for families and host regular conferences.
- STXBP1 Disorders Foundation Page: https://www.stxbp1disorders.org
- STXBP1 Summit+ 2022 Session Recordings: https://www.stxbp1disorders.org/2022-stxbp1-summit-recordings/#ResearchRoundtableRecording
Research studies. The scientific community is actively studying STXBP1 and its role in human disease. The STXBP1 Epilepsiome team is happy to facilitate if you have questions or a specific interest in this gene.
References
Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. Jun 2008;40(6):782-8. doi:10.1038/ng.150
Stamberger H, Nikanorova M, Willemsen MH, et al. STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology. Mar 8 2016;86(10):954-62. doi:10.1212/WNL.0000000000002457
Xian J, Parthasarathy S, Ruggiero SM, et al. Assessing the landscape of STXBP1-related disorders in 534 individuals. Brain. Jun 3 2022;145(5):1668-1683. doi:10.1093/brain/awab327
Abramov D, Guiberson NGL, Burre J. STXBP1 encephalopathies: Clinical spectrum, disease mechanisms, and therapeutic strategies. J Neurochem. Jul 8 2020;doi:10.1111/jnc.15120
O’Brien S, Ng-Cordell E, Study DDD, Astle DE, Scerif G, Baker K. STXBP1-associated neurodevelopmental disorder: a comparative study of behavioural characteristics. J Neurodev Disord. Aug 6 2019;11(1):17. doi:10.1186/s11689-019-9278-9
Otsuka M, Oguni H, Liang JS, et al. STXBP1 mutations cause not only Ohtahara syndrome but also West syndrome–result of Japanese cohort study. Epilepsia. Dec 2010;51(12):2449-52. doi:10.1111/j.1528-1167.2010.02767.x
Hamdan FF, Piton A, Gauthier J, et al. De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann Neurol. Jun 2009;65(6):748-53. doi:10.1002/ana.21625
Carvill GL, Weckhuysen S, McMahon JM, et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. Apr 8 2014;82(14):1245-53. doi:10.1212/WNL.0000000000000291
Verhage M, Sorensen JB. SNAREopathies: Diversity in Mechanisms and Symptoms. Neuron. Jul 8 2020;107(1):22-37. doi:10.1016/j.neuron.2020.05.036
Lammertse HCA, van Berkel AA, Iacomino M, et al. Homozygous STXBP1 variant causes encephalopathy and gain-of-function in synaptic transmission. Brain. Feb 1 2020;143(2):441-451. doi:10.1093/brain/awz391
Kovacevic J, Maroteaux G, Schut D, et al. Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. Brain. May 1 2018;141(5):1350-1374. doi:10.1093/brain/awy046
Guiberson NGL, Pineda A, Abramov D, et al. Mechanism-based rescue of Munc18-1 dysfunction in varied encephalopathies by chemical chaperones. Nat Commun. Sep 28 2018;9(1):3986. doi:10.1038/s41467-018-06507-4
Milh M, Villeneuve N, Chouchane M, et al. Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia. Oct 2011;52(10):1828-34. doi:10.1111/j.1528-1167.2011.03181.x
Balagura G, Xian J, Riva A, et al. Epilepsy Course and Developmental Trajectories in STXBP1-DEE. Neurol Genet. Jun 2022;8(3):e676. doi:10.1212/NXG.0000000000000676
Sullivan KR, Ruggiero SM, Xian J, et al. A disease concept model for STXBP1-related disorders. Epilepsia Open. Jan 10 2023;doi:10.1002/epi4.12688