
SNARE complex. This is the first post in our series on SNAREopathies – an umbrella term for diseases caused by variants in the soluble NSF attachment protein receptor (SNARE) complex, that is essential for neuronal synaptic vesicle exocytosis in the presynapse. The core SNARE complex comprises a four-helix bundle consisting of two SNAP25 helices, which is encoded by SNAP25, the syntaxin 1 helix encoded by STX1A/STX1B, and the synaptobrevin 2 helix encoded by VAMP2. The complex is regulated by different proteins including MUNC18 encoded by STXBP1, MUNC13 encoded by UNC13A, and synaptotagmin encoded by SYT1 and complexins. Here is an overview of SNAREopathies, with a focus in this post on SNAP25 and VAMP2.
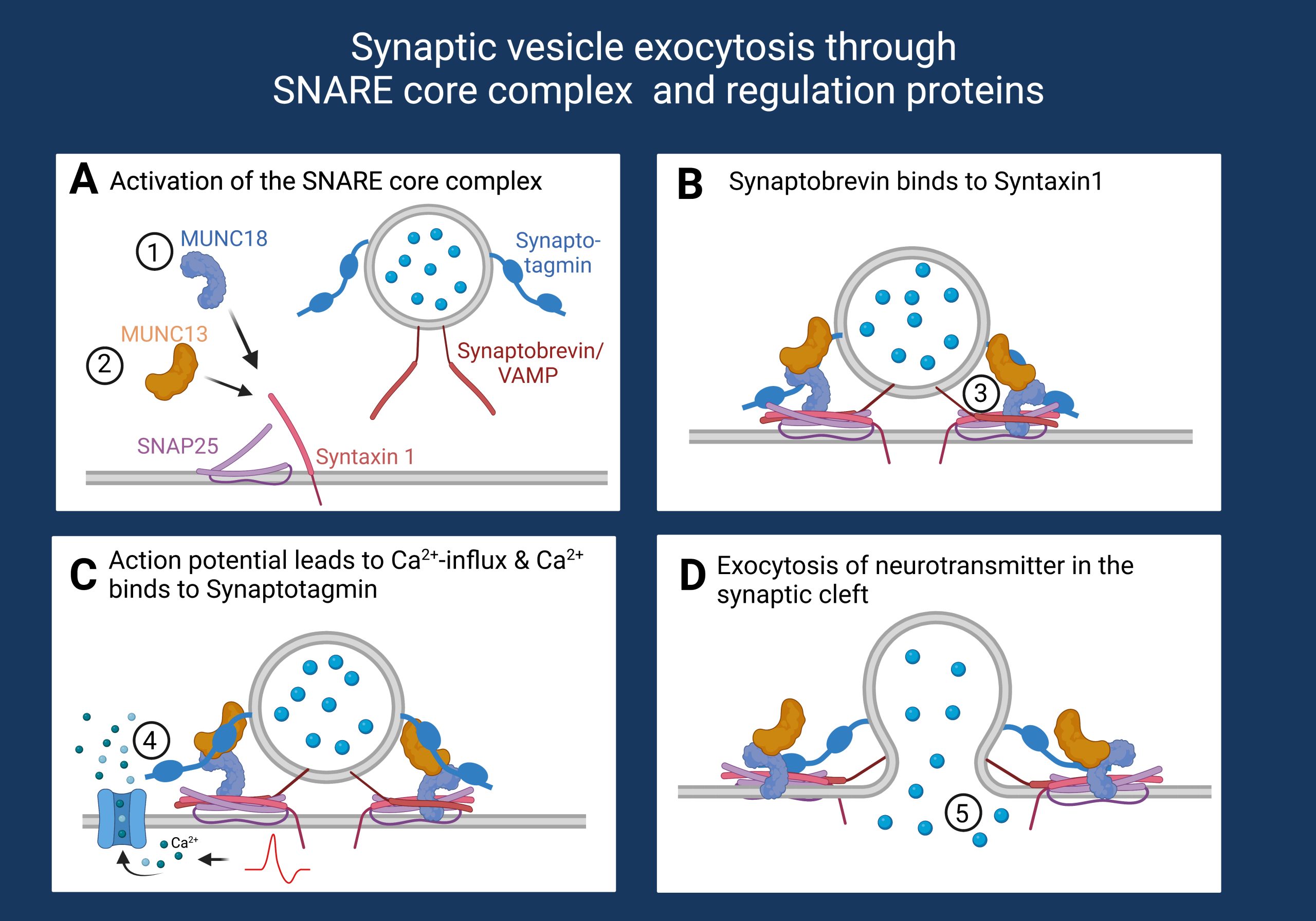
Figure 1. Synaptic vesicle release is coordinated through the SNARE complex. 1. Binding of MUNC18 leads to a closed confirmation of Syntaxin-1. 2. Binding of MUNC13 opens the Syntaxin-1 confirmation and allows binding to SNAP25. 3. Synaptobrevin binds to the t-SNARE proteins leading to the formation of the SNARE complex. 4. An arriving action potential leads to calcium influx in the presynapse and calcium binds to Synaptotagmin. 5. Neurotransmitter exocytosis in the synaptic cleft. Created with BioRender.
Phenotypes. Inactivation of one of the genes encoding for a SNARE-complex protein leads to decreased vesicle exocytosis. In humans, disease-causing variants in genes coding for each of the SNARE-complex proteins, referred to as “Snareopathies,” lead to neurodevelopmental phenotypes with or without epilepsy. Even though all SNARE- complex proteins work together to achieve vesicle exocytosis, phenotypes related to variation in each of the genes are distinct. In our series on SNAREopathies in epilepsies and neurodevelopmental disorders, we wanted to provide you with an overview of recent advances in understanding the genotypic and phenotypic landscape of SNAREopathies. Here, we start our series with two genes encoding proteins that form the core SNARE complex: SNAP25 and VAMP2.
SNAP25 mechanism. SNAP25 is associated with the nerve terminal membranes – t-SNARE protein – and enables synaptic vesicle fusion by pairing with proteins that are incorporated into the membranes of the transport vesicles, known as v-SNARE proteins. SNAP25 is essential for neuronal survival. De novo and heterozygous variants in SNAP25 have been reported to cause disease, including 15 different missense variants and four variants associated with a loss-of-function disease mechanism. Recurrent variants include p.Gly43Arg, p.Met71Thr, and p.Gln174*. The variant I67T/N has been functionally tested and found to have a dominant-negative effect.
SNAP25 phenotypes. So far, 23 individuals with SNAP25-related disorders have been described, with profound DD and/or ID and early-onset seizures as the main symptoms (Klöckner et al., 2021). DD/ID were present in all individuals, ranging from profound (20%) to mild (5%). Most individuals were able to walk and two third of individuals communicated verbally. 74% of individuals had seizures, with onset primarily before two years of age. Seizures in SNAP25 were characterized by generalized or focal to bilateral tonic–clonic seizures, absence-like seizures, and epileptic spasms. Notably, half of individuals with epilepsy had intractable seizures. Other phenotypic features included muscular hypotonia, spasticity, movement disorders, cerebral visual impairment (CVI), and musculoskeletal findings. Behavioral issues were reported to be less common. The SNAP25 foundation was founded in 2020 with the goal to advocate for affected families and to raise money to support research.
VAMP2 mechanism. The VAMP2 gene encodes synaptobrevin-2, the only vesicular-binding protein (v-SNARE) of the SNARE core complex and the most numerous of proteins on a synaptic vesicle. The interaction with t-SNARE proteins via formation of a four-helix bundle leads to the binding of the synaptic vesicle with the presynaptic cell membrane. The genotypic spectrum includes seven missense variants, two indels, one nonsense variant, and one frameshift variant, all localized in the highly conserved SNARE motif. All variants have been reported as de novo and heterozygous.
VAMP2 phenotypes. Only eleven individuals with disease causing variants in VAMP2 have been described to date. All individuals had DD and/or ID. Half of individuals had seizures with mainly generalized onset. Of these individuals with seizures, half had focal-onset seizures and/or infantile spasms. Similarly, as with SNAP25-related disorders, other features included muscular hypotonia, movement disorders, and CVI. In contrast to SNAP25-related disorders, behavioral features in VAMP2 were reported in all individuals.
What you need to know. The SNARE-complex is essential for presynaptic vesicle exocytosis. Even though all proteins are involved in the same process, phenotypes of affected individuals both overlap and are distinct. Individuals with disease causing variants in SNAP25, encoding the t-SNARE protein present with mild to profound ID and early-onset seizures. All individuals with disease causing variants in VAMP2, encoding the only v-SNARE protein Synaptobrevin-2, present with DD and/or ID and behavioral features and seizures were present in half of the individuals. Given the increasing recognition of SNAREopathies, the genetic and phenotypic characterization of core proteins SNAP25 and VAMP2 is critical in understanding the role of synapse-related genes in epilepsies and neurodevelopmental disorders.
This post was co-written by Kim Marie Thalwitzer and Julie Xian.