
Rett. We have written very little about MECP2 on Beyond the Ion Channel. MECP2 is the gene for Rett Syndrome, a neurodegenerative disorder almost exclusively affecting females. Classical Rett Syndrome is characterized by developmental regression in the first two years of life and the development of distinctive hand movements, which historically led to Rett Syndrome being considered a recognizable entity. This blog post is the introduction to our MECP2 Epilepsiome page. However, in 2016, a time when many genes are re-defined by exome studies, I was wondering whether Rett Syndrome is still the classical syndrome that I initially learned about.
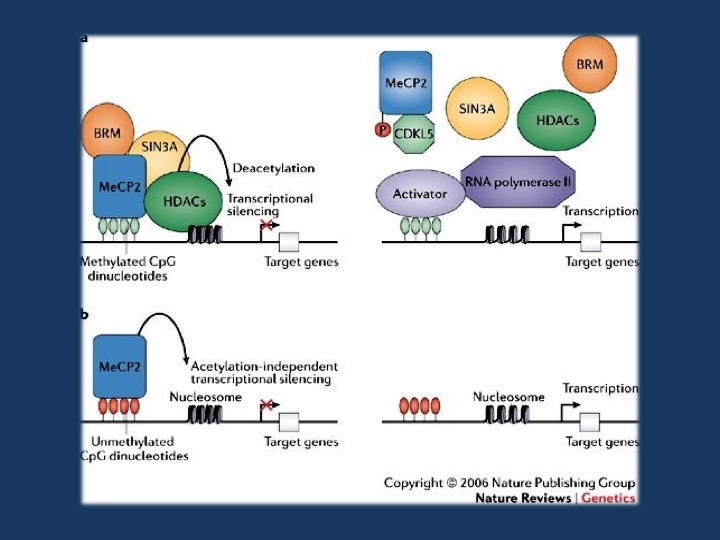
Figure 3 from Bienvenu & Chelly, 2006. a | Transcription is suppressed in promoter regions containing methylated CpGs that are bound by MeCP2 (methyl-CpG-binding protein 2). MeCP2 binds methylated DNA and recruits chromatin-remodeling complexes that contain SIN3A (a transcriptional co-repressor), BRM (a SWI/SNF-related chromatin-remodelling protein) and histone deacetylases (HDACs). When MeCP2 is not bound to methylated DNA (right panel), the complex that usually contains MeCP2, BRM, SIN3A and HDACs is not recruited. This could result from a missense mutation in the methyl-CpG-binding domain or loss of expression of MeCP2, for example, because of an early-truncating mutation (these scenarios are not shown here). In each of these cases, histones remain acetylated and the DNA at the promoter remains in an open conformation, allowing transcription factors to bind DNA and initiate transcription. b | MeCP2 is also a potent chromatin-condensing protein and can repress gene expression independently of DNA methylation, at least in vitro71 (left panel). At promoters where this DNA-methylation-independent function of MeCP2 is involved in regulating expression, a deficiency or absence of MeCP2 leads to a disorganization of chromatin structure (indicated here by increased spacing between nucleosomes), making transcription more likely to occur (right panel). Reprinted by permission from Macmillan Publishers Ltd: Nature Review Genetics (Bienvenu T., Chelly J. 2006. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 7, 415–426. 10.1038/nrg1878), copyright 2006.
Epilepsiome. The MECP2 Epilepsiome page was written by our genetic counselling student Kathleen Kenley in collaboration with Elizabeth Dechene, who has coordinated our Epilepsiome posts for the last year. The reason why I wanted to point out their effort is the fact that we are actively trying to recruit professionals into our Epilepsiome project that will slowly fuse with the NIH-funded ClinGen initiative. With her debut post, Kathleen has taken on one of the neurodevelopmental genes that we have somewhat neglected on Beyond the Ion Channel. Even though more than half of the patients with Rett Syndrome have epilepsy, it was a gene that rarely came up during our posts on gene discovery in epilepsy. We have mentioned genes for Rett-related disorders frequently, including CDKL5 and FOXG1. MECP2, however, has rarely been mentioned.
The phenotypic spectrum. Our impression of so many genes has changed dramatically through exome studies. When you have the possibility to go from genotype to phenotype rather than being limited to testing genes based on clinical features alone, you sometimes discover a wider phenotypic spectrum than you had imagined. And the width of the phenotypic spectrum of certain genes is nothing than can be predicted. WDR45, for example, has transitioned from a gene for a rare iron storage disorder, to a gene with a phenotype that is so broad that more patients are identified through exome studies than through targeted testing. Other disorders, however, such as many recessive metabolic disorders, rarely have atypical presentations. Where on this spectrum does MECP2 stand in 2016?
MECP2 in 2016. Atypical presentations of Rett Syndrome are frequently found. For example, in our publication of 293 epilepsy patients that had undergone exome sequencing, variants in MECP2 in addition to KCNQ2 were the most common cause to be found. Also, MECP2 comes up in many other studies when patients with unclear phenotypes undergo exome sequencing. It is not a gene for epileptic encephalopathies like many of the other genes that we frequently discuss on our blog, but it is a gene that is regularly identified in patients with unclear epilepsy phenotypes. This observation alone tells us that it is sometimes important to re-think what we consider conceptual boundaries between disease entities. Rett Syndrome is a clinical phenotype that is probably much broader than we initially anticipated.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
