
GLUT1DS. This is the Epilepsiome page for SLC2A1, coding for the major glucose transporter in brain and erythrocytes. Initially thought to be exclusively associated with a very rare and severe epileptic encephalopathy, recent studies have suggested that SLC2A1 mutations can also be identified in patients with early-onset generalized epilepsies and familial epilepsies.
Here are the most recent blog posts on SLC2A1
- The glucose transport bottleneck – This is what you need to know about SLC2A1 in 2015
- Publications of the week: CDKL5, KIF1A, and familial cortical tremor
- Identifying the Doose gene – SLC6A1 mutations in Myoclonic Astatic Epilepsy
In a nutshell. GLUT1 Deficiency Syndrome was first described in 1991 by De Vivo and collaborators. The initial description of this condition referred to two children with global developmental delay, seizures and an unexplained low glucose in their cerebrospinal fluid, a condition referred to as hypoglycorrhachia. Seven years later, the gene underlying this condition was identified as SLC2A1. Ever since the initial description, both dominant and recessive forms have been identified, and the phenotypic range has expanded to movement disorders and particular types of epilepsy.
Phenotypes | Genetics | Mechanism | Community
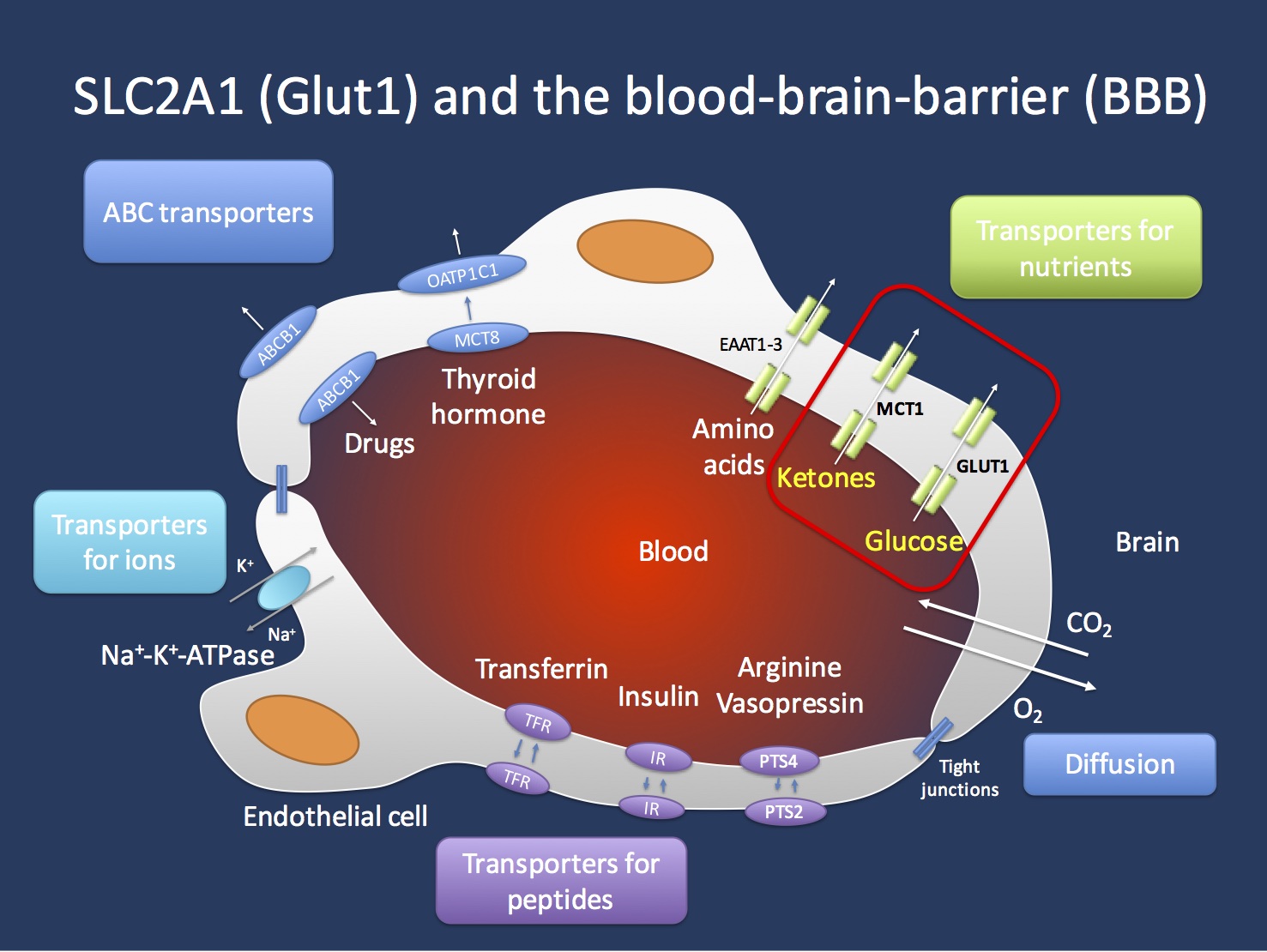
Transport across the blood brain barrier (BBB). The endothelial cells in the Central Nervous System are important gatekeepers of transport into the CNS. The GLUT1 transporter encoded by the SCL2A1 gene is the main transporter for glucose and mutations in the SLC2A1 gene lead to GLUT1 Deficiency Syndrome. This condition can be treated with the ketogenic diet that allows the brain to primarily use ketones as an alternate source of energy. Ketones are primarily transported through the MCT1 transporter, which is encoded by the SLC16A1 gene. Figure inspired by Zlokovic, Nature Reviews Neuroscience 12, 723-738 (December 2011).
Phenotype
GLUT1DS. There are various phenotypes associated with mutations in SCL2A1. Classical GLUT1DS is associated with severe epilepsy starting in infancy, microcephaly, and difficult to treat epilepsy. A major shift in our thinking about SLC2A1 occurred when mutations in this gene were identified in early-onset absence epilepsy (EOAE), a generalized epilepsy with early and often refractory onset of absence seizures. The cut-off between early-onset absences and typical absences is arbitrarily set at 4 years, even though some authors use different cut-offs. Patients with EOAE often have learning difficulties and many have intellectual disability. More recently, Myoclonic Astatic Epilepsy (MAE) has been suggested as a second epilepsy phenotype due to SLC2A1 mutations. In families of probands with SCL2A1-related generalized epilepsies, the mutation has been found to segregate with various epilepsy phenotypes including focal epilepsies. The discovery of SCL2A1 mutations in up to 10% of EOAE and MAE patients has raised the question to what extent patients with typical Idiopathic/Genetic Generalized Epilepsy (IGE/GGE) have GLUT1DS. Even though it may still be possible that a very small subset of typical IGE/GGE patient carry causative mutations in SLC2A1, it is very uncommon finding. Likewise, GLUT1DS appears to be a very rare cause of epileptic encephalopathies on a population basis. In the Epi4K/E2 cohort of more than 350 patients with Infantile Spasms or Lennox-Gastaut-Syndrome, not a single patients with GLUT1DS was identified through exome sequencing. GLUT1DS can be treated with the ketogenic diet. Given these treatment implications, some clinicians recommend either genetic or metabolic testing for GLUT1DS in patients with atypical, refractory epilepsy, particularly if the patient has generalized epilepsy with early onset.
The non-epileptic phenotypes of GLUT1DS are intellectual disability and paroxysmal exercise-induced dystonia. There is some debate whether non-syndromic intellectual disability or autism can be due to GLUT1DS, but there is currently little data to support this notion.
Genotype
Haploinsufficiency. GLUT1DS is a genetically homogeneous condition and mutations in SCL2A1 are the only genetic cause for this condition. Other than two families who have been described with recessive GLUT1DS, most patients have heterozygous mutations in the SCL2A1 gene. Deletion, truncations, frameshift mutations or splice site mutations are thought to lead to a more severe phenotype such as classical GLUT1DS, which is characterized by early-onset epilepsy, global developmental delay, movement disorders, and microcephaly. In milder cases, such as the patients with EOAE and MAE, missense mutations have been observed. To my knowledge, no truncation mutations have been found in patients with generalized epilepsy.
Resources. There is a very comprehensive GeneReviews section on Glut 1 deficiency syndrome that was updated in January 2015. However, I feel that this review focuses more on the classical severe phenotype and not so much on the diverse milder epilepsy syndromes associated with SCL2A1 mutations. The OMIM page on SCL2A1 is very comprehensive, listing many of the established references. Also, the NORD database entry is very detailed and up-to-date.
I have a patient with a variant in SCL2A1. What does this mean? If you’re holding a genetic result indicating a variant in SLC2A1 in your hands, you have already made the decision that the patient has a genetic epilepsy that requires a genetic work-up. How does the positive result of a pathogenic mutation change your clinical management? In contrast to many other genetic epilepsy, GLUT1DS has a biomarker, the CSF/serum glucose ratio, which is typically 0.5 or higher. Therefore, you can confirm the possible diagnosis in CSF. The SLC2A1 gene is not very large and false positive findings on exome or gene panel analysis are rarely encountered compared to other genetic epilepsies. Clinical features such as movement disorders, fluctuations of symptoms during the day with worsening shortly before meals, and paradoxical worsening during illnesses would prompt you making the diagnosis. However, given the wide phenotypic range and potential treatment implications, confirmatory work-up may even be considered in atypical phenotypes. Again, particularly early-onset generalized epilepsies including EOAE would prompt you to seek such a work up. A PET scan or glucose uptake assay in erythrocytes can be used as additional tests, but these tests are not frequently used in routine clinical practice to make the diagnosis of GLUT1DS.
Mechanism
Transport. GLUT1DS is a one gene – one molecular function – one mechanism disease. Glucose transport across the blood brain barrier (BBB) is a tightly regulated system that requires sufficient amounts of the GLUT1 transporter to present on the endothelial cells, which line the brain capillaries. In patients with GLUT1 deficiency, the transport across the BBB is insufficient to provide sufficient amounts of glucose to the brain, resulting in a chronic CNS glucose starvation. The presence of sufficient GLUT1 transporters is a true bottleneck in the supply of the CNS with nutrients. GLUT1 facilitates diffusion across the BBB in an energy-independent mechanism. This means that the transport of glucose across the endothelial cells is only dependent on the amount of GLUT1 transporters present on the endothelium. Some older studies suggest that brain endothelial cells transport 10x their weight of glucose per minute. The particular effects of chronic insufficient glucose transport on molecular and cellular changes in the CNS are not well understood. Interestingly, this transporter defect may generate generalized discharges as we would usually see in genetic ion channel disorders. It is currently unexplained why a chronic shortage of glucose transport across the BBB may result in such a phenotype, but it may indicate a specific vulnerability of the thalamocortical circuitry that is implicated in generating generalized discharges. GLUT1DS can be effectively treated with the ketogenic diet. In brief, the CNS can use ketones as an alternative source of energy and ketones are transported across the BBB through a different transporter, the MCT1 transporter that is encoded by the SLC16A1 gene.
Community
The Glut1 Deficiency Foundation maintains an active website with Glut1-related patient information.