
SCN8A. This is the Epilepsiome page for SCN8A, encoding the voltage-gated sodium channel alpha subunit Nav1.6, which has been implicated in early infantile epileptic encephalopathies, as well as other epilepsy phenotypes.
Here are the most recent blog posts on SCN8A
- SCN8A – this is what you need to know in 2015
- SCN8A encephalopathy – and how it differs from Dravet Syndrome
- Publications of the week: GRIN2A, SCN8A, and DEPDC5
- Publications of the week: SCN8A, SYN1, ZDHHC9, and SCNM1
In a nutshell. SCN8A was first implicated in epilepsy in 2012. Since then, more than 100 patients with a phenotypic spectrum ranging from benign infantile seizures and paroxysmal dyskinesia to severe early-infantile epileptic encephalopathies (EIEE13) caused by heterozygous SCN8A mutations have been identified. The distinguishing features of SCN8A-associated EIEE3 are the infrequency of febrile seizures and the movement disorders, ranging from mild ataxia to choreoathetosis and even to quadriplegia. The predominant mechanism underlying EIEE13 appears to be neuronal hyperexcitability caused by gain of function variants.
| Phenotypes | Genetics | Mechanism & Function |
| The Clinical Perspective | Community | Resources & References |
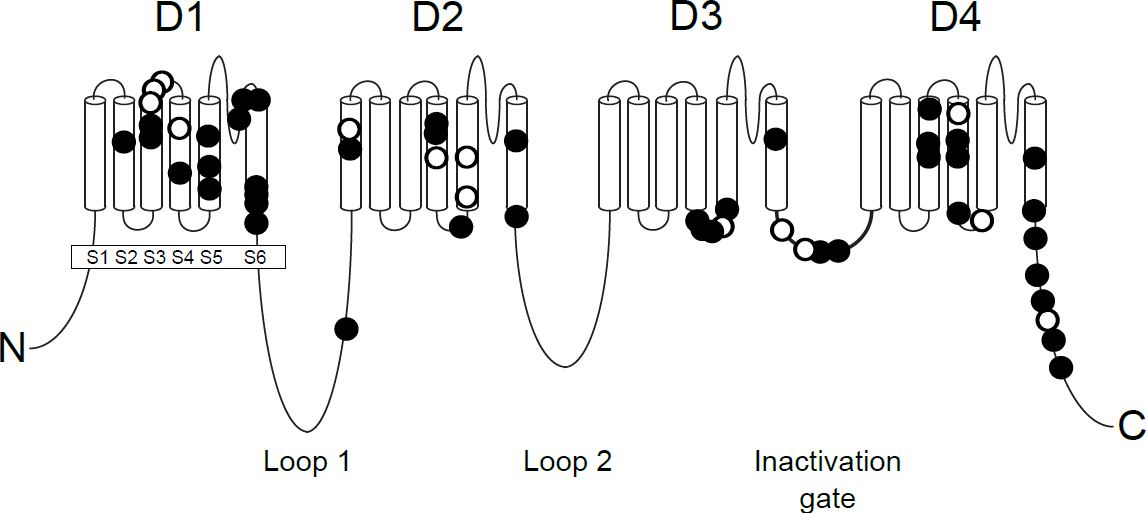
SCN8A encodes the voltage-gated sodium channel alpha subunit Nav1.6. Adapted from Wagnon and Meisler, Front Neurol 2015.
Phenotypes
Early Infantile Epileptic Encephalopathy 13 (EIEE13). SCN8A epileptic encephalopathy is characterized by seizure onset between birth and 18 months of age (mean of 5 months), mild to severe intellectual disability, and developmental delay with or without regression. Seizures are frequently polymorphic. Generalized tonic-clonic seizures occur in many patients, and tonic, atonic, myoclonic, focal and absence seizures are not uncommon.
In contrast with Dravet syndrome, febrile seizures are rarely observed in EIEE13. EEG findings in patients with EIEE13 may be normal at the time of seizure onset but often comprises moderate to severe background slowing with focal or multifocal epileptiform discharges after seizure onset.
In addition to seizures, motor manifestations, such as ataxia and choreoathetosis, are common and hypotonia, hypertonia, and/or dystonia are present in approximately half of the patients with EIEE13. In some cases, the patients cannot sit or walk unassisted. Many patients have very little to no speech, and some patients gradually lose eye contact during the course of the disease. SUDEP (sudden unexpected death in epilepsy) has been reported in 12% of cases.
Benign infantile seizures and paroxysmal dyskinesia. Sixteen affected family members from 3 unrelated families were identified to carry an identical heterozygous SCN8A missense mutation (p.E1483K). All affected individuals presented with benign familial infantile seizures (BFIS) during the first years of life. Interictal EEG, cognition, and development of motor skills were normal in the majority of the cases. Thirty percent of affected family members had single unprovoked seizures later in life. One third of the patients developed paroxysmal dyskinetic/dystonic movements in puberty, triggered by stretching, motor initiation or by emotional stimuli. This variant did not seem be due to a founder effect (Gardella et al 2015).
Intellectual disability. SCN8A variants have also been identified in individuals with intellectual disability without a history of epilepsy.
Genetics
Mutation Spectrum (Type and Location). More than 50 mutations in SCN8A have been reported in patients with early-infantile epileptic encephalopathy (EIEE13, OMIM 614558). The majority of variants associated with EIEE13 and benign infantile seizures and paroxysmal dyskinesia are missense variants except for one splice site mutation that causes an in-frame deletion and an intragenic deletion. Recently, three loss-of-function (LOF) variants of SCN8A have also been associated with epilepsy. Two missense variants caused protein instability or loss of channel activity and were associated with EIEE, and one deletion of exons 2-14 was found in a patient with early-onset absence epilepsy. Approximately 30% of disease-causing variants are recurrent.
Most pathogenic mutations of SCN8A are located in the transmembrane domains of the affected protein, as explained below. Details of all the known pathogenic variants will be available soon in an online SCN8A mutation database (see Community below).
General Considerations for Variant interpretation. When reviewing a genetic variant to determine its significance for a given patient, it is important to weigh multiple pieces of evidence:
Gene level interpretation. First, it is important to establish the strength of the evidence showing that the gene is associated with epilepsy. Some genes may only have one variant reported in a single individual with epilepsy, while other genes may have multiple variants reported in many large families with an autosomal dominant pattern of epilepsy.
Variant level interpretation. When reviewing the significance of a variant, it is important to consider the impact on the gene and the presence of the variant in previously described patient and control populations. Many clinical genetic testing laboratories classify genetic variants into different categories, ranging from benign to pathogenic. Variants that are common in control populations and would not be predicted to have a major impact on the gene/protein are generally classified asbenign. Variants are more likely to be classified as pathogenic if the variants are rare or not present in the control population, reported in multiple individuals or families with disease, and likely to have a higher impact on the gene/protein based on the type of mutation or functional studies. Variants with uncertain or limited available evidence may be classified as variants of uncertain significance (VUS), indicating that further information is required in order for the variant to be further defined. In some cases, testing additional family members can be helpful, as it allows the lab to determine whether or not the variant was inherited (versus de novo) and how the variant segregates with disease in the family. Sometimes further classification of a VUS requires waiting for the identification of additional patients or families with similar or nearby variants.
Inheritance, Penetrance & Prevalence.
SCN8A-associated epilepsies are automosal dominant disorders, and thus affected individuals are heterozygous for the SCN8A variant. In most cases with EIEE13, the mutation arises de novo in the patient. In three EIEE13 cases, the variant was inherited from a mosaic parent who was unaffected. The small number of families identified to have an SCN8A variant associated with benign infantile seizures and paroxysmal dyskinesia all had an autosomal dominant pattern of disease. Thus far, SCN8A-associated conditions are believed to have complete penetrance with variable expressivity. On four large screens of individuals with epileptic encephalopathies, de novo mutations of SCN8A were identified in 1% of cases (13/1557).
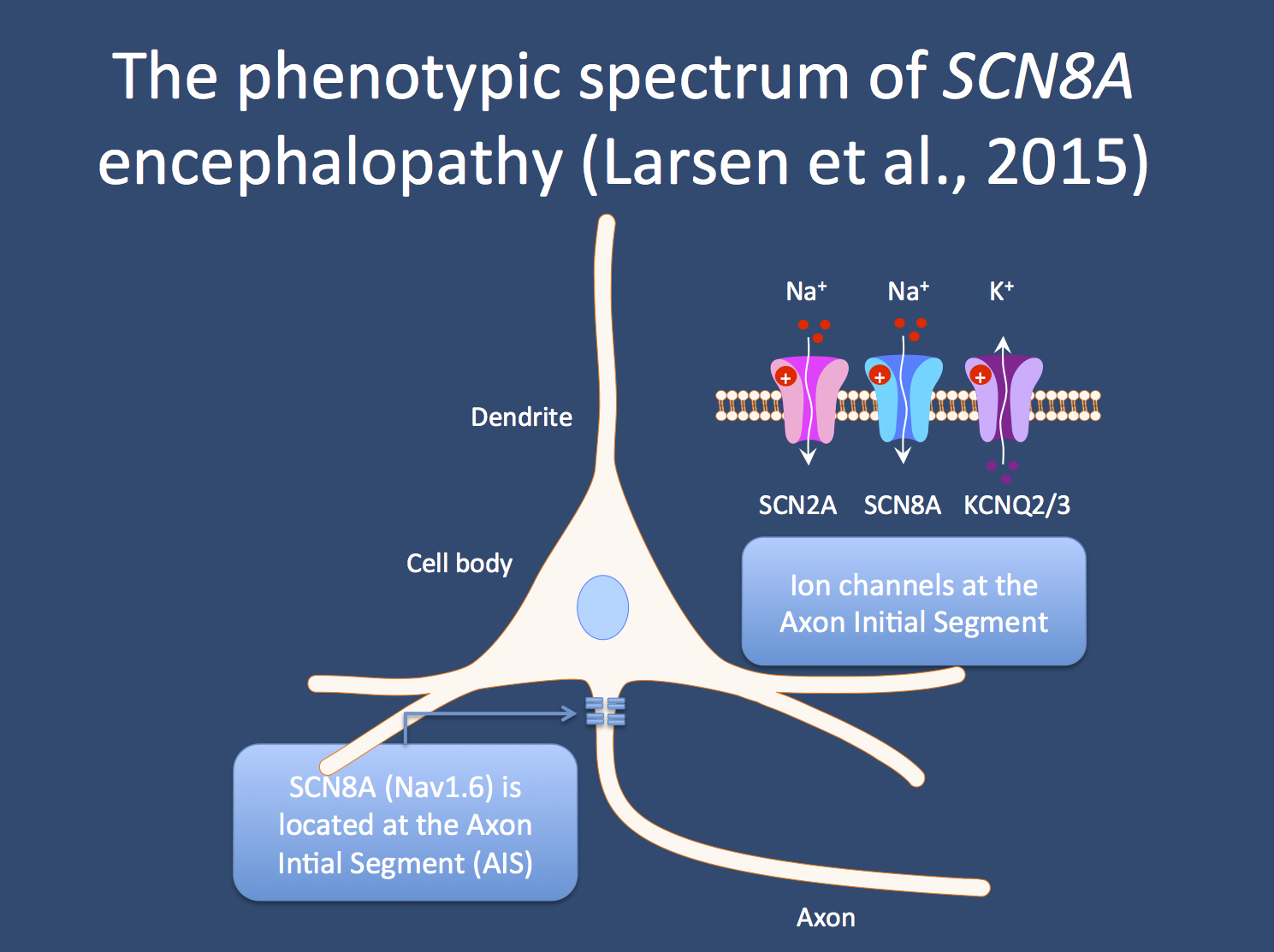
The ion channels encoded by the SCN2A, SCN8A, and KCNQ2 genes are located at the axon initial segment, the part of the neuron where all excitatory and inhibitory impulses at the neuronal membrane are integrated and translated into an action potential. Mutations in all three genes are now associated with severe epilepsies. For SCN2A and SCN8A, the mutations are assumed to be excitatory; for KCNQ2, the mutations are thought to be dominant negative.
Mechanism & Function
Nav1.6. SCN8A encodes the voltage-gated sodium channel alpha subunit Nav1.6. The Nav1.6 protein comprises 4 homologous domains that each have 6 transmembrane segments, two large intracellular loops, the inactivation gate located between D3 and D4, and intracellular N- and C-terminal domains (See Figure). The pathogenic mutations are predominantly located in the transmembrane domains, the inactivation gate, and the C-terminus. These regions of the channel are much more highly conserved through evolution compared to the less well-conserved large intracellular loops and are more likely to be involved in channel function.
Neuronal voltage-gated sodium channels regulate cellular excitability by controlling the flow of sodium ions across the cell membrane. Nav1.6 is expressed in excitatory and inhibitory neurons in the brain. Nav1.6 is localized in high concentrations at the axon initial segments of neurons and at nodes of Ranvier where it mediates action potential initiation and propagation.
Hyperexcitability. Ten EIEE13-associated variants have been tested functionally. Eight of the 10 demonstrate gain-of-function (GOF) features, including hyperpolarizing shifts in the voltage-dependence of activation, impaired inactivation, and elevated persistent current. The GOF features cause the Nav1.6 channel to be hyperactive, which leads to increased neuronal firing. These findings indicates that neuronal hyperexcitability caused by GOF mutations is the predominant mechanism underlying EIEE13 (Wagnon et al, 2015). The recurrent variant associated with benign infantile seizures and paroxysmal dyskinesia has not yet been tested functionally.
Different from Dravet. This GOF mechanism is distinct from the mechanism underlying Dravet syndrome, a key differential diagnosis for EIEE13. More than 85% of Dravet syndrome cases are caused by disease-causing variants in the related sodium channel SCN1A. The majority of SCN1A variants in Dravet syndrome cause loss-of-function of the sodium channel Nav1.1. Thus, medications that further reduce sodium channel activity (e.g. carbamazepine, lamotrigine) are contraindicated in Dravet syndrome. However, in EIEE13, these medications can reduce or prevent seizures in some patients because they may reduce the elevated sodium channel activity observed with EIEE13 mutations.
Cell/Animal models.
Mice. Multiple mouse models of SCN8A-associated disease have been reported. These mice have a variety of levels of impact on SCN8A functioning, ranging from complete lack of SCN8A function to gain-of-function variants. The mice have variable phenotypes. Mice with loss of function or reduced function due to homozygous mutations in SCN8A have movement abnormalities, including ataxia, tremor, muscle weakness and dystonia as seen in some individuals with SCN8A variants. Mice with heterozygous loss of function variants have less severe phenotypes, such as spike-wave discharges suggestive of absence epilepsy, disrupted sleep architecture, and behavioral abnormalities such as anxiety (Summarized in O’Brien et al, 2013).
A heterozygous knock in mouse model expressing a dominant gain-of-function variant (p.Asn1768Asp) was found to present with seizures and sudden unexpected death in epilepsy, similar to the phenotype in humans. The mice exhibit mildly impaired motor coordination and social discrimination prior to the onset of seizures. The seizure phenotype has incomplete penetrance. Homozygous mice have earlier onset of gait abnormalities and impaired motor coordination, followed quickly by seizure onset and seizure-induced death.
The Clinical Perspective
Recurrence Risk. All established disease-causing variants associated with EIEE3 identified thus far appear to be de novo or sporadic. Although not yet reported, germline mosaicism of CHD2 mutations is possible, making the recurrence risk for families with one child with an SCN8A variant associated with EIEE3 higher than the general population risk.
SCN8A variants associated with benign infantile seizures and paroxysmal dyskinesia are inherited in an autosomal dominant pattern. Therefore, each child of an individual with a disease-causing variant has a 50% of inheriting the disease-causing variant and an equal chance of inheriting the functional (wild-type) copy of the gene.
Therapy. Seizures are generally refractory to treatment, but sodium channel blockers including carbamazepine, oxcarbazepine, and phenytoin have been used successfully in several patients.
Community
Online Registry. An online registry for SCN8A families is available at www.scn8a.net. The website also provides resources for families, clinicians, and researchers. Two non-profit organizations, Wishes for Elliott: Advancing SCN8A Research (www.wishesforelliott.org) and The Cute Syndrome Foundation (www.thecutesyndrome.com), provide information about SCN8A epileptic encephalopathy and support SCN8A research.
Research studies. The scientific community is currently actively studying SCN8A and its role in human disease. The SCN8A Epilepsiome team is happy to facilitate if you have questions or a specific interest in this gene.
Resources & References
Boerma RS et al. 2016. Remarkable phenytoin sensitivity in 4 children with SCN8A-related epilepsy: a molecular neuropharmacological approach. Neurotherapeutics 13(1):192-7. PMID: 26252990
Gardella E et al. 2015. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol. Epub ahead of print. PMID: 26677014
Larsen J et al. 2015. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015;84:480–489. PMID: 25568300
Ohba et al. 2014. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 55(7):994-1000. PMID: 24888894
O’Brien JE and Meisler MH. 2013. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet 4:213. PMID: 24194747
Veeramah KR et al. 2012. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 90(3):502-10. PMID: 22365152
Wagnon et al. 2015. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet. 24(2): 506–15. PMID: 25227913
Wagnon JL and Meisler MH. 2015. Recurrent and non-recurrent mutations of SCN8A in epileptic encephalopathy. Front Neurol 6:104. PMID: 26029160
Wagnon et al. 2015. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol. 3(2):114-23. PMID: 26900580