
ACMG. Imagine the following scenario: you identify a de novo variant in SCN1A in a young child with the typical clinical features of Dravet Syndrome. However, the lab returns the variant as a variant of uncertain significance. The variant is a missense variant that has never been seen before and the lab argues that they are simply applying the current variant classification criteria. Certainly, either the lab is wrong or the variant classification criteria are deficient. Shouldn’t this variant be a pathogenic variant? Your patient clearly has the typical clinical features that are very unlikely explained by anything but the de novo SCN1A variant. In fact, both assumptions are incorrect, but it is important to know the background. Here is a blog post on why variant classification is distinct from assessing whether variants are explanatory in a clinical context. And please allow me to introduce a neologism: explanatoriness.
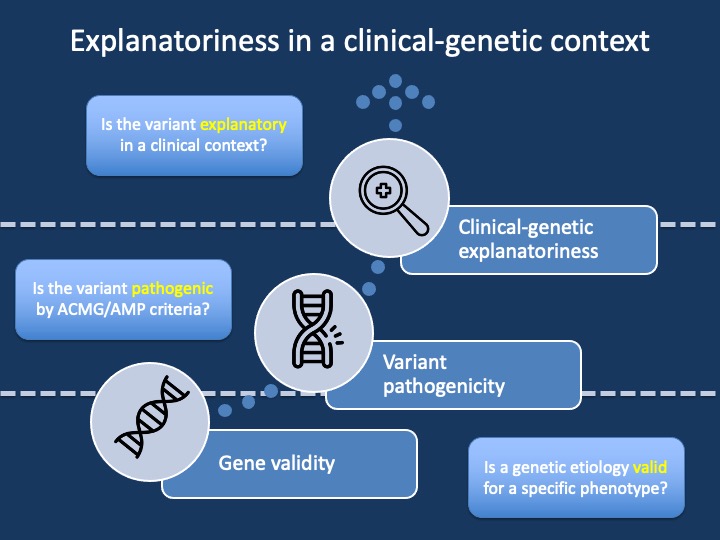
Figure 1. When trying to understand whether a specific genetic variant explains an individual’s phenotype, we have typically separated this assessment into two distinct questions. The first question is whether the gene (more precisely the “genetic etiology”) is valid for the overall phenotype. For example, CACNA1H is a valid genetic etiology for early-onset hypertension, but not for epilepsy. This is referred to as assessing the gene-disease validity. Once gene-disease validity has been established, the next step is the assessment of the specific variant in the gene. This classification is referred to as variant pathogenicity. However, these two assessments do not provide a final diagnosis. All variants require a clinical correlation and an assessment of whether an identified variant in a valid genetic etiology is explanatory in a clinical context. This blog post is dedicated to this third, clinical level of making a genetic diagnosis – assessing the “explanatoriness” of a variant.
ACMG. When we took on the role of the variant classification criteria for epilepsy genes two years ago, we had lofty goals. The official variant classification criteria are generic with much room for improvement. I hoped that we could transfer much of our clinical epilepsy genetic knowledge to the field of variant classification, thereby reducing the number of uncertain variants (VUS) and allowing us to make earlier genetic diagnoses. However, I realized that I had a conceptual misunderstanding of how variant classification works. For example, our draft revised variant classification for epilepsy-related sodium channels includes a wide range of additional criteria that will make variant classification more specific: functional data, critical domains, paralogue variants, stricter cut-offs for population frequencies, and so on. However, with the exception of a few limited scenarios, one major aspect is largely missing: phenotypes.
Paradox #1. At first glance, it seems like a major oversight that phenotypic features are not included. The phenotype for Dravet Syndrome is very specific and recognizable. So why aren’t these features included? It took me almost two years to understand it, but here is the short answer: phenotypes cannot be included as the ACMG/AMP criteria are variant criteria. A patient phenotype is not a property of a genetic variant in the same way that a population frequency or CADD score is. This might be become clearer with the following example: consider a large GEFS+ family with an inherited SCN1A variant as we reported in 2013. The phenotypes in the affected family member range widely and some individuals are even unaffected. Would this variant be pathogenic in only affected individuals and benign in unaffected individuals?
Paradox #2. Also, imagine a scenario where you assess the coding region and identify an SCN1A missense variant in an individual with typical Dravet Syndrome. If the individual’s phenotype were to be included in the classification, this variant would potentially become pathogenic. However, let’s suppose that the individual’s phenotype is actually caused by a deep intronic variant rather than the missense variant. We can never certainly exclude all other causes for an individual’s phenotype. Accordingly, variant pathogenicity must come from the property of each variant. Assessing whether the variant fits with an individual’s phenotype must therefore happen OUTSIDE of ACMG criteria. While this might sound surprising given the effort that we put into improving the ACMG criteria, this framework is what we typically apply to other testing modalities. We would never assume that an MRI image or EEG report would be an individual’s final diagnosis. All these tests require correlation in a clinical context. However, for genetic variants, we are hesitant to give this process a name.
Explanatoriness. A few years ago, a new phrase made it into my patient letters. I started to include a statement describing whether or not we think that an identified variant is explanatory in the context of an individual’s phenotype. For example, the SCN1A de novo missense variant mentioned above cannot be more than a VUS on the testing report (i.e., it cannot be considered likely pathogenic or pathogenic), but it would be considered explanatory in a clinical context. Likewise, we encountered likely pathogenic variants that we dismissed as non-explanatory in the past. Let me claim a new term for this concept: explanatoriness. We need to assess a gene’s validity, a variant’s pathogenicity, and the explanatoriness of the clinical-genetic context [in case you’re wondering, it’s explanatoriness, not explainability; explainability refers to the fact whether the features of a system can be sufficiently explained. In contrast, explanatoriness, while sounding odd at first, refers to the quality of being explanatory – it is a proper English word].
Context. The concept of explanatoriness is neither surprising nor revolutionary. Putting testing modalities into clinical context is commonplace in clinical care. And there are often limitations to what an MRI, CT, or EEG can show. Accordingly, the epilepsy genetics field has three rather than two tasks to improve precision medicine through precision diagnosis. We need to become more certain about gene-disease relationships (gene validity), improve our capacity for ascertaining which variants are pathogenic and which are not (variant pathogenicity), and we have to develop concepts for how we tackle the explanatoriness of a variant given an individual’s phenotype. Importantly, introducing the new concept should not introduce a subjective component into making a genetic diagnosis. Whether a variant is explanatory can be governed by the same principles that we use for gene validity and variant pathogenicity.
SCN1A. What would such an assessment look like? For example, in the scenario mentioned above, we could try to arrive at a reasonable estimate on how likely it is that a de novo missense variant is explanatory given the individual’s phenotype. You would probably agree with me that this probability is relatively high. While hidden intronic variants or other, non-sequenced genes may be causative, most experts would probably agree that this probability of this kind of undetected variant is low, and the chance therefore of the identified variant being causative is 90% or higher. Contrast this with a novel missense variant in CACNA1A without parental testing in an individual with new-onset infantile spasms. Yes, this variant could also theoretically be causative, but the likelihood is much lower. However, both variants are VUS. Only a framework for clinical correlation could differentiate between the near certainty of the de novo SCN1A variant and the uncertainty of the CACNA1A variant.
Exception. If you are reading this as an expert in variant curation, you might be scratching your head. Yes, there actually is a way to include phenotypic information in variant curation, the so-called case-counting method. For recurrent variants, the ACMG criteria allow us to score the phenotypes of individuals with recurrent variants and the phenotype of the patient in front of you can add to this classification. However, this case is a fringe example. Most recurrent variants will likely become pathogenic because of their recurrence, the case-counting method has traditionally not applied to novel variants (even though we’re in the process of addressing this). Therefore, while this exception is a unique way to introduce phenotypes into the ACMG criteria, it is not a generally applicable framework for clinical-genetic correlation.
Quantifying phenotypes. The task for the future is to formalize a framework to assess whether identified variants are explanatory in a clinical context and base these assumptions on actual numbers rather than intuition. Invariably, this leads us to an important question: how do we measure phenotypes? For example, if we had a framework to compare phenotypes across a large patient cohort, we could use the phenotypes of individuals with known pathogenic variants to assess how much more likely an identified SCN1A variant is to be causative rather than non-causative. It will come down to probabilities and evidence based on large patient cohort. Luckily, frameworks for this kind of analysis are emerging. We have worked with the Human Phenotype Ontology (HPO) in the last five years, which has allowed us to archive and digitize phenotypes for entire patient cohorts, such as all reported individuals with SCN2A or STXBP1. Similar projects are currently on the way for a range of other genetic epilepsies, including SYNGAP1, CACNA1A, SCN8A, and others. We hope that by making phenotypes quantifiable, we can make meaningful steps towards developing frameworks to assess how likely genetic variants are to be explanatory for our patients’ phenotypes.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
