
MECP2. This is the Epilepsiome page for MECP2, which is associated with Rett syndrome, MECP2 duplication syndrome, MECP2-related severe neonatal encephalopathy and PPM-X syndrome [OMIM#300005]. Occurring almost exclusively in females, Rett syndrome is characterized by developmental regression, generalized tonic-clonic seizures and complex partial seizures. MECP2-related severe neonatal encephalopathy primarily affects males and presents with encephalopathy and severe seizures. Caused by duplication of the entire MECP2 gene, MECP2 duplication syndrome occurs almost exclusively in males and is characterized by moderate to severe intellectual disability, spasticity, absence seizures and generalized tonic-clonic seizures. PPM-X syndrome primarily affects males and presents with psychotic disorders, intellectual disability and movement disorders, but so far has not been associated with seizures.
Here are the most recent blog posts on MECP2
- Three novel aspects about epilepsy gene panels
- The novel gene dilemma
- CDKL5 – this is what you need to know in 2015
- How genome sequencing in intellectual disability breaks the 50% boundary
In a nutshell. Over a decade later, MECP2 variants were identified in some cases of Rett syndrome. Soon after, the NIH completed a natural history study of Rett syndrome, which confirmed the importance of consistent application of diagnostic criteria as treatment trials were developed. In 2000, Meloni and colleagues reported on the first males identified to have MECP2 variants and found that they were not able to survive past a year of age. This report was followed by a rapid increase of reported males with MECP2-related conditions, including males with whole gene duplications.
| Phenotypes | Genetics | Mechanism & Function |
| The Clinical Perspective | Community | Resources & References |
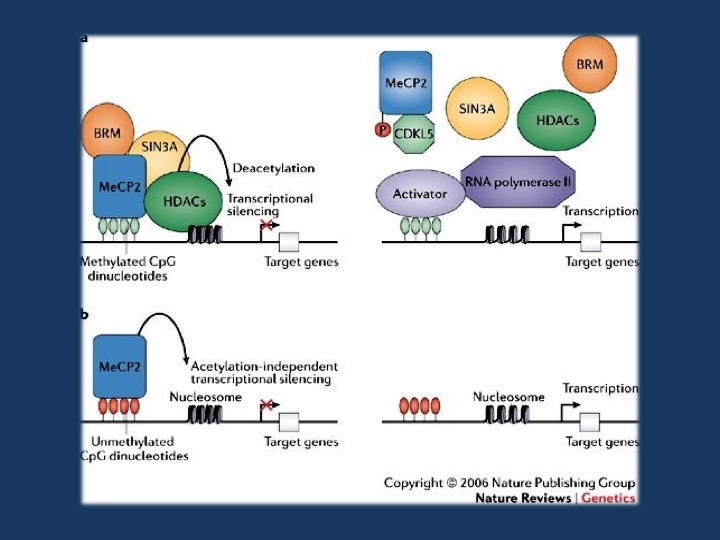
Figure 3 from Bienvenu & Chelly, 2006. a | Transcription is suppressed in promoter regions containing methylated CpGs that are bound by MeCP2 (methyl-CpG-binding protein 2). MeCP2 binds methylated DNA and recruits chromatin-remodeling complexes that contain SIN3A (a transcriptional co-repressor), BRM (a SWI/SNF-related chromatin-remodelling protein) and histone deacetylases (HDACs). When MeCP2 is not bound to methylated DNA (right panel), the complex that usually contains MeCP2, BRM, SIN3A and HDACs is not recruited. This could result from a missense mutation in the methyl-CpG-binding domain or loss of expression of MeCP2, for example, because of an early-truncating mutation (these scenarios are not shown here). In each of these cases, histones remain acetylated and the DNA at the promoter remains in an open conformation, allowing transcription factors to bind DNA and initiate transcription. b | MeCP2 is also a potent chromatin-condensing protein and can repress gene expression independently of DNA methylation, at least in vitro71 (left panel). At promoters where this DNA-methylation-independent function of MeCP2 is involved in regulating expression, a deficiency or absence of MeCP2 leads to a disorganization of chromatin structure (indicated here by increased spacing between nucleosomes), making transcription more likely to occur (right panel). Reprinted by permission from Macmillan Publishers Ltd: Nature Review Genetics (Bienvenu T., Chelly J. 2006. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 7, 415–426. 10.1038/nrg1878), copyright 2006.
Phenotypes
Rett Syndrome. The classical clinical picture of Rett Syndrome is characterized by normal psychomotor development during the first 6 to 18 months of life, followed by a rapid regression in language and motor skills. Up to 90% of females with Rett syndrome have generalized tonic-clonic seizures and complex partial seizures. Seizure frequency is greatest when the disease stabilizes after the regression stage. EEG findings for individuals with Rett syndrome show slowing of the occipital dominant rhythm and background activity with spike or sharp wave discharges during sleep. Patients may have episodes of apnea and hyperventilation, laughing, screaming, and vacant staring spells. Individuals with Rett syndrome also have repetitive movements, spasticity, feeding difficulties, which can lead to progressive growth failure and wasting, as well as bowel dysmotility.
MECP2 duplication syndrome is characterized by moderate to severe intellectual disability, spasticity, increased susceptibility to infections, absence seizures and generalized tonic-clonic seizures. These seizures may not improve with treatment. EEG findings in individuals with MECP2 duplication are the same as the EEG findings in individuals with Rett syndrome.
MECP2-related severe neonatal encephalopathy is characterized by encephalopathy, hypotonia, movement disorders, rigidity and seizures. Males with this condition often pass away before the age of 3 due to respiratory failure.
PPM-X is characterized by psychotic disorders (most commonly bipolar disorder), intellectual disability and movement disorders in males. The majority of affected males only present with intellectual disability. Most carrier females are unaffected, but mild intellectual disability has been observed in a few carrier females. It is unclear how variants in the MECP2 gene lead to the signs and symptoms of PPM-X syndrome.
Genetics
Mutation spectrum. Over 500 different disease-causing variants have been reported in the scientific literature. The most common variants are missense variants and deletions, but duplications and complex rearrangements have also been identified. Disease-causing variants have been reported throughout the gene. To view some of the variants that have been reported thus far, please see the ClinVar page for MECP2.
Genotype-Phenotype Correlation. In females, pathogenic missense variants and deletions of the MECP2 gene are associated with classic Rett syndrome in the majority of cases. Missense variants and deletions seen in live-born males are associated with MECP2-related severe neonatal encephalopathy and PPM-X syndrome. Duplications of the MECP2 gene are associated with MECP2 duplication syndrome. Discordance in genotype and expected phenotype have been reported which is thought to be due to epigenetic or other genetic factors contributing to the phenotype.
General Considerations for Variant interpretation. When reviewing a genetic variant to determine its significance for a given patient, it is important to weigh multiple pieces of evidence:
Considerations for gene level interpretation. First, it is important to establish the strength of the evidence showing that the gene is associated with epilepsy. Some genes may only have one variant reported in a single individual with epilepsy, while other genes may have multiple variants reported in many large families with an autosomal dominant pattern of epilepsy. For MECP2, there is moderate evidence for its role in human epilepsy.
Considerations for variant level interpretation. When reviewing the significance of a variant, it is important to consider the impact on the gene and the presence of the variant in previously described patient and control populations. Many clinical genetic testing laboratories classify genetic variants into different categories, ranging from benign to pathogenic. Variants that are common in control populations and would not be predicted to have a major impact on the gene/protein are generally classified as benign. Variants are more likely to be classified as pathogenic if the variants are rare or not present in the control population, reported in multiple individuals or families with disease, and likely to have a higher impact on the gene/protein based on the type of mutation or functional studies. Variants with uncertain or limited available evidence may be classified as variants of uncertain significance (VUS), indicating that further information is required in order for the variant to be further defined. In some cases, testing additional family members can be helpful, as it allows the lab to determine whether or not the variant was inherited (versus de novo) and how the variant segregates with disease in the family. This is particularly true for MECP2 variants, as most disease-causing variants would be expected to be de novo. Sometimes further classification of a VUS requires waiting for the identification of additional patients or families with similar or nearby variants.
Inheritance, Penetrance & Prevalence. MECP2 is located on the X chromosome, and, therefore, MECP2-related conditions are inherited in an X-linked pattern. The majority of cases are single occurrences in a family due to a de novo pathogenic variant or due to a parent with germline mosaicism. However, there are reported cases of a mother having a MECP2 pathogenic variant and being unaffected due to skewed X-chromosome inactivation. The prevalence of Rett syndrome is estimated to be 1 in 8,500 females. The prevalence of MECP2 duplication syndrome and MECP2-related severe neonatal encephalopathy is unknown.
Mechanism & Function
Mechanism. The MECP2 gene encodes for a protein called methyl-CpG binding protein 2 (MeCP2), which is thought to help regular gene activity by modifying chromatin. MeCP2 protein is present in cells throughout the body, but is particularly abundant in brain cells where it plays a role in maintaining connections between neurons. Recent research has led to the hypothesis that Rett syndrome is a disorder of synaptic plasticity and that agents that correct this imbalance may have beneficial effects on the brain development in these patients.
The Clinical Perspective
Recurrence risk & testing of family members. MECP2 variants can be de novo or inherited in an X-linked pattern. Each child of a mother who carries a disease-causing variant has a 50% (1 in 2) of inheriting the disease-causing variant. It is appropriate to test a seemingly unaffected mother of an affected proband for the same disease-causing variant due to skewed X-inactivation. If the disease-causing variant is not identified in the mother, the risk to future pregnancies is low, but germline mosaicism of MECP2 variants has been reported, making the recurrence risk for families with one child with a MECP2 variant higher than the general population risk.
Therapy. Treatment of MECP2-related conditions is focused on symptoms and optimizing the individual’s abilities. Treatment for seizures needs to be individualized, but individuals with MECP2 duplication syndrome may not have seizures improve from treatment. Individuals with Rett syndrome may benefit from risperidone to treat agitation and melatonin to ameliorate sleep disturbances.
The current section on therapy is a brief overview, and, from the therapeutic perspective, does not represent a comprehensive review of the literature. It should therefore not be used for clinical decision-making without input from a health care provider with relevant expertise in the field.
Community
Research studies, patient registries, and family connections. The Epilepsiome team is happy to facilitate if you have questions or a specific interest in this gene. There are a number of groups/websites that provide relevant information and support for individuals and families with MECP2-related conditions, including the groups listed below.
- American Epilepsy Society
- Epilepsy Foundation
- Epilepsy Action UK
- Rett Syndrome.org
- National Organization for Rare Disorders (NORD)
- MECP2 Duplication Syndrome
Resources & References
Resources
For Clinicians:
GeneReviews: MECP2-Related Disorders
Orphanet: Rett Syndrome
For Families:
NIH Genetics Home Reference on MECP2-related epilepsy