
WDR45. This is the Epilepsiome page for WDR45, a gene initially identified as a causative disease gene for a rare group of neurodegenerative iron storage disorders referred to as neurodegeneration with brain iron accumulation (NBIA). Ever since the initial discovery, the phenotypic range of WDR45 has significantly broadened. Mutations in WDR45 can be identified in a wide range of phenotypes, including in patients with intellectual disability and epileptic encephalopathies. WDR45 is an X-chromosomal gene and phenotypes observed in males are thought to be more severe compared to females.
Here are the most recent blog posts on WDR45
- WDR45 – this is what you need to know in 2015
- Publications of the week: Genetic attribution and stigma, SCN2A, WDR45
- The eye of the tiger, brain iron and the beta-propeller
- The 30-10 rule of clinical exome sequencing
- How genome sequencing in intellectual disability breaks the 50% boundary
In a nutshell. WDR45 is a candidate gene for a broad range of neurodevelopmental and neurodegenerative disorders. Historically, WDR45 mutations have been identified in patients with static encephalopathy with neurodegeneration in adulthood (SENDA). Given the widespread use of genetic testing, mutations in WDR45 are often identified in patient before sign of iron storage can be identified. The phenotype of patients with WDR45 mutations through genetic testing can be broad with affected males more severely affected than females.
Phenotypes | Genetics | Mechanism | Community
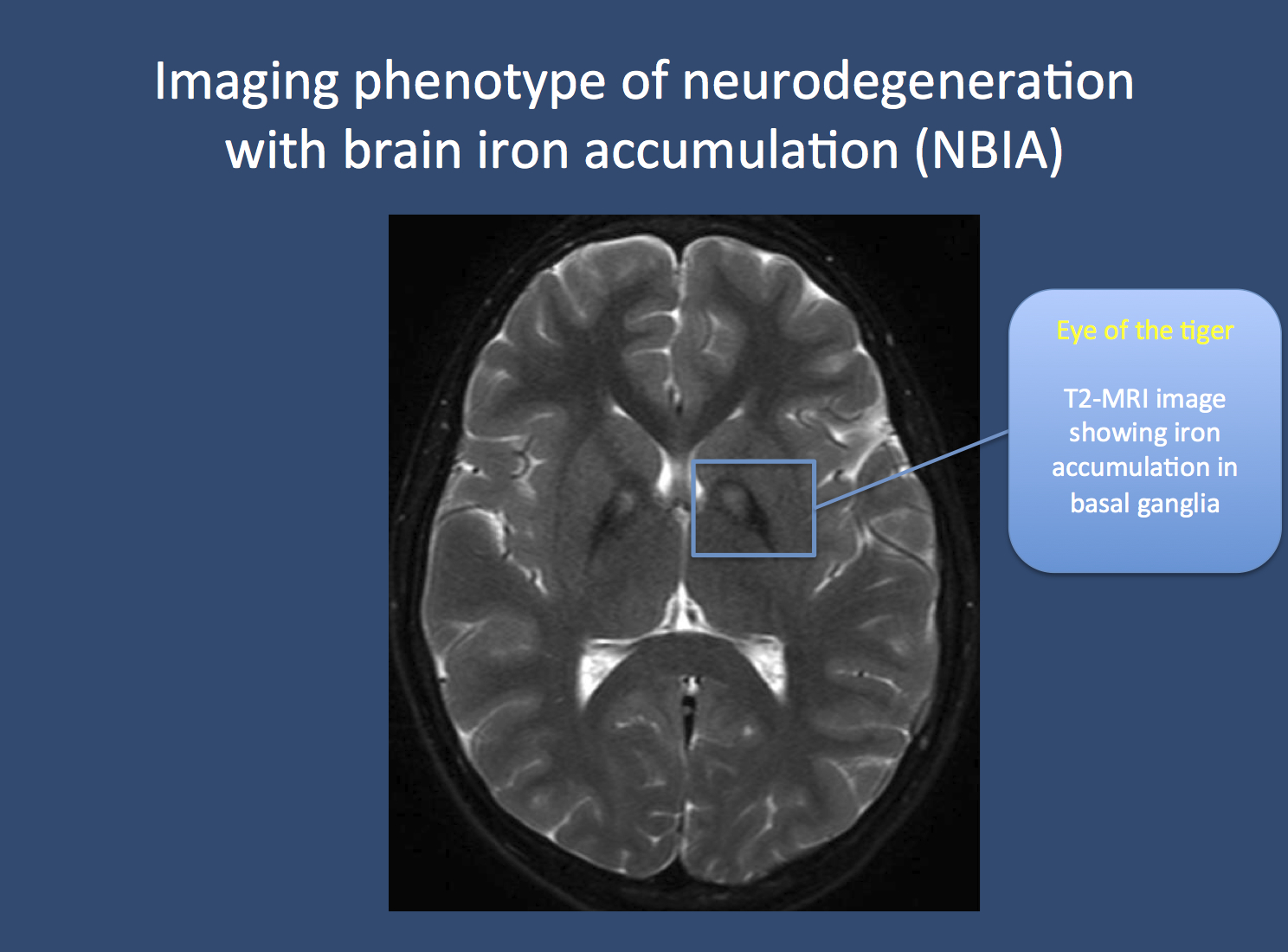
Figure. The eye of the tiger in NBIA in a patient with pantothenate kinase-associated neurodegeneration (PKAN). Neuroimaging in patients with PKAN and other NBIA is usually associated with a pathognomonic finding. Due to the accumulation of iron in the basal ganglia, two black spots can be seen, which is referred to as the “eye of the tiger” sign. PKAN is part of a group of disorders referred to as neurodegeneration with brain iron accumulation. Image from Wikimedia commons (http://commons.wikimedia.org/wiki/File:Pkan-basal-ganglia-MRI.JPG), within the framework of the Creative Commons license.
Phenotypes
Overview. The phenotypic spectrum of patients with WDR45 mutations is broad, ranging from mild cognitive impairment in females to severe early-onset epileptic encephalopathies in males. In some patients with mild and relative static cognitive issues, there is a worsening in adulthood with the onset of dystonia and dementia. We have distinguished four different, albeit overlapping phenotypes associated with WDR45.
SENDA. Static encephalopathy with neurodegeneration in adulthood (SENDA) is the classical phenotype associated with mutations in WDR45. This condition, which was initially observed by Haack and collaborators in 2012 and Saitsu and collaborators in 2013, refers to patients presenting with an initially non-progressive and “static” intellectual disability with subsequent development of neurodegenerative features in early adulthood that comprise dementia and dystonia. SENDA may be related to a phenotype of intellectual disability with early-onset parkinsonism reported by Nishioka and collaborators.
BPAN. Some females with WDR45 mutations have phenotypes reminiscent of Rett Syndrome or atypical Rett Syndrome. Given the properties of the WDR45 protein, Hayflick and collagues opted to call the various phenotypes associated with WDR45 mutations beta-propeller associated neurodegeneration (BPAN). The authors felt that “static encephalopathy” did not entirely capture the early presentation of the disease. In the early phase of the disease, patients were found to have global developmental delay, epilepsy and stereotypies, i.e. repetitive movements such as hand flapping. Particularly the latter reminded the authors of other diseases including Rett Syndrome and atypical Rett Syndrome. The epilepsy phenotypes comprised myoclonic, astatic and absence seizures. Furthermore, the authors suggest that neuroimaging might be normal in childhood. In young adulthood, dystonia, parkinsonism and progressive dementia were noted and the MRI showed basal ganglia iron deposits.
Intellectual disability. There are various reports in the literature of females with intellectual disability, but no other features such as epilepsy or movement disorders. Some of these patients were identified through exome sequencing. It is not clear at this point to what extent these patients have been followed over a longer period of time to assess whether their phenotype is stable or whether there were features of SENDA.
Epileptic encephalopathies. Some male patients with early-onset epileptic encephalopathies and de novo mutations in WDR45 have been identified. Abidi and collaborators have identified a male patient with WDR45 mutation and early-onset refractory seizures who was found to have a de novo WDR45 truncating mutation.
Genetics
Truncating mutations. The various diseases associated with WDR45 mutation are typically found in association with truncating de novo mutations in WDR45. This suggests that the underlying genetic mechanism is haploinsuffiency. Most patients with de novo WDR45 mutations are females and it is assumed that mutations in males are often lethal prenatally. The few male patients with de novo truncating mutations have a severe phenotype with early-onset seizures and profoundly delayed neurological development. The vast majority of mutations in WDR45 are de novo truncating mutations. A family with an inherited WDR45 mutation was reported by Zarate and collaborators. In this family, an inframe deletion was transmitted from a mother with a mosaic mutation to a son with intellectual disability and seizures and a less severely affected daughter who did not have seizures. BPAN is the only NBIA subtype that is X-linked.
Mechanism
Beta-propeller. A beta-propeller is a protein that is made out different protein sheets that are arranged around a central tunnel comparable to propeller-blades. WDR45 is one of these proteins, and it is assumed that the beta-propeller is a scaffold that serves as a platform for protein–protein interactions that are important in autophagy, the removal of residual cellular components. It remains unclear how haploinsufficiency in WDR45 is connected to human disease.
Community
Community. The NBIA disorders association has an active website and connects families, medical professionals, and researchers.