
CACNA1H. This is the Epilepsiome page on CACNA1H, a gene initially considered to be linked to absence epilepsies. However, the initial publications on the role of CACNA1H have not been supported by more recent studies. CACNA1H may play a role in a different group of diseases, namely early-onset hypertension due to primary aldosteronism.
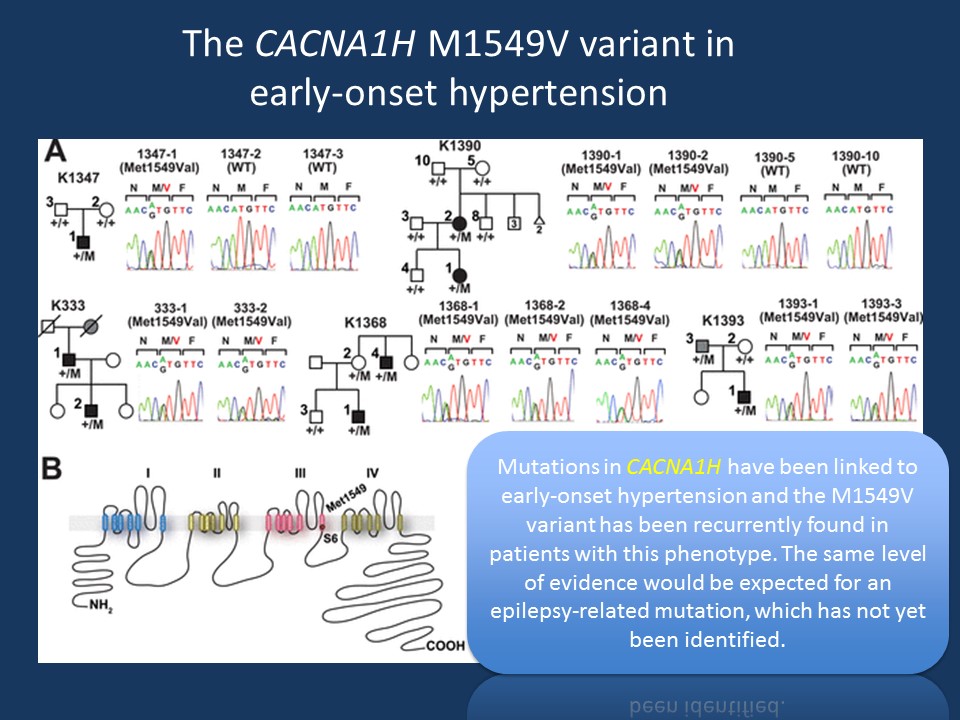
Figure modified from Scholl and collaborators under a Creative Commons Attribution license. Figure legend modified from Scholl and collaborators. (A) Pedigrees of kindreds with CACNA1H M1549V mutation. Studied subjects with early-onset hypertension are shown as black filled symbols, and subjects with early-onset hypertension by family history (K333) or low renin with normal blood pressure (K1393) are shown as grey filled symbols. (B) Transmembrane structure of CaV3.2 (encoded by CACNA1H), the pore-forming subunit of a voltage-gated Ca2+ channel, is shown. These channels have four internal homologous repeats (I–IV), each with six transmembrane segments (S1–S6) and a membrane-associated loop between the pore-forming S5 and S6 segments. The p.Met1549Val mutation is located in S6 of repeat III (modified from http://elifesciences.org/content/4/e06315 under a Creative Commons Attribution license)
Here are the most recent blog posts on CACNA1H
- CACNA1H – this is what you need to know in 2015
- EFHC1 – retiring an epilepsy gene
- CACNA2D2, the ducky mouse, and what it takes to be an epilepsy gene
- Beyond the Ion Channel – and back
In a nutshell. CACNA1H is an epilepsy candidate gene that is mainly thought to be a risk factor for absence epilepsies. Using the criteria that we usually apply to either genes for monogenic diseases or for genetic risk factors, there is currently insufficient evidence to link variation in the CACNA1H gene to human epilepsy.
Phenotype | Genotype | Mechanism | Community
Phenotype
Absence Epilepsy. The initial publications on CACNA1H suggested a role for rare variants as risk factors in childhood absence epilepsies (CAE). More recent studies investigating common variants and rare variants do not support a role of CACNA1H variants in CAE. None of the initially reported variants were shown to be de novo or segregating with the disease.
Primary aldosteronism. Rather unexpected, recurrent mutations in CACNA1H have recently been described in early-onset hypertension and primary aldosteronism, an endocrinological disorder. CACNA1H is expressed in the adrenal gland and the mutations were shown to be gain-of-function. In total, Scholl and collaborators identified 5 patients with the identical M1549V mutation, which was shown to be de novo in two patients. None of the patients had epilepsy or neurodevelopmental disorders.
Genotype
CACNA1H in databases. Here is brief overview of what is known about CACNA1H in genome databases. The CACNA1H gene is one of the genes in the human genome that tolerates heterozygous loss-of-function mutations either due to deletions (DGV) or due to loss-of-function mutations (ExAC). Apart from the recurrent M1549V mutation identified in early-onset hypertension, two de novo mutations have been identified in patients with autism and one de novo mutation was identified in an unaffected sibling. We would assume at this point that all de novo mutations other than the recurrent M1549V mutation are incidental findings unrelated to disease.
Epilepsy risk factors. Is it possible that variants in CACNA1H are risk factors for genetic epilepsies and may in fact contribute to disease that we are missing at this point? Yes, but it is difficult for us to make a case for this at this point. We can never say never. All we can say is that we currently do not have the technologies or sample sizes to track down a small effect of CACNA1H variants in human epilepsies. However, the current studies do not provide evidence that the role of CACNA1H is particularly strong.
Mechanism
Hyperexcitability. CACNA1H was initially of interest to the epilepsy community because it codes for a thalamic T-type calcium channel. T-type calcium channels are critical in regulating the excitability level of thalamic pacemaker neurons, which are thought to be involved in the pathogenesis of 3-Hz spike-and-wave associated with generalized absence seizures. The lines of evidence linking CACNA1H to epilepsy in animal models is also indirect. In contrast to other mouse models with calcium channel mutation such as stargazer (CACNG2), lethargic (CACNB4) and ducky (CACNA2D2), CACNA1H mouse models do not present with epilepsy.
Community
There is currently no research community or patient organization for patients carrying CACNA1H variants.