
SYNGAP1. This is the Epilepsiome page of SYNGAP1, a gene for generalized developmental and epileptic encephalopathy. SYNGAP1 was initially discovered in 2009 as non-syndromic intellectual disability (ID) gene. Since then, SYNGAP1 has established itself as a prominent monogenic cause for ID, generalized epilepsy, global developmental delay, and autism spectrum disorder.
Here are the most recent blog posts that mention SYNGAP1:
- SYNGAP1 – this is what you need to know
- Closing the knowledge gap – this is SYNGAP1
- AES 2022 – Soundbites from Nashville, Tennessee
- Walking in Memphis – TARGETing Epilepsy at St. Jude
- Unlocking STXBP1 through Electronic Medical Records
In a nutshell. SYNGAP1 encodes a regulatory protein of the postsynaptic density that is involved in neuronal plasticity. De novo pathogenic variants in SYNGAP1 have been observed in patients with non-syndromic intellectual disability, autism, and patients with epileptic encephalopathy, mainly with generalized seizure types.
Phenotypes | Genetics | Mechanism | Community
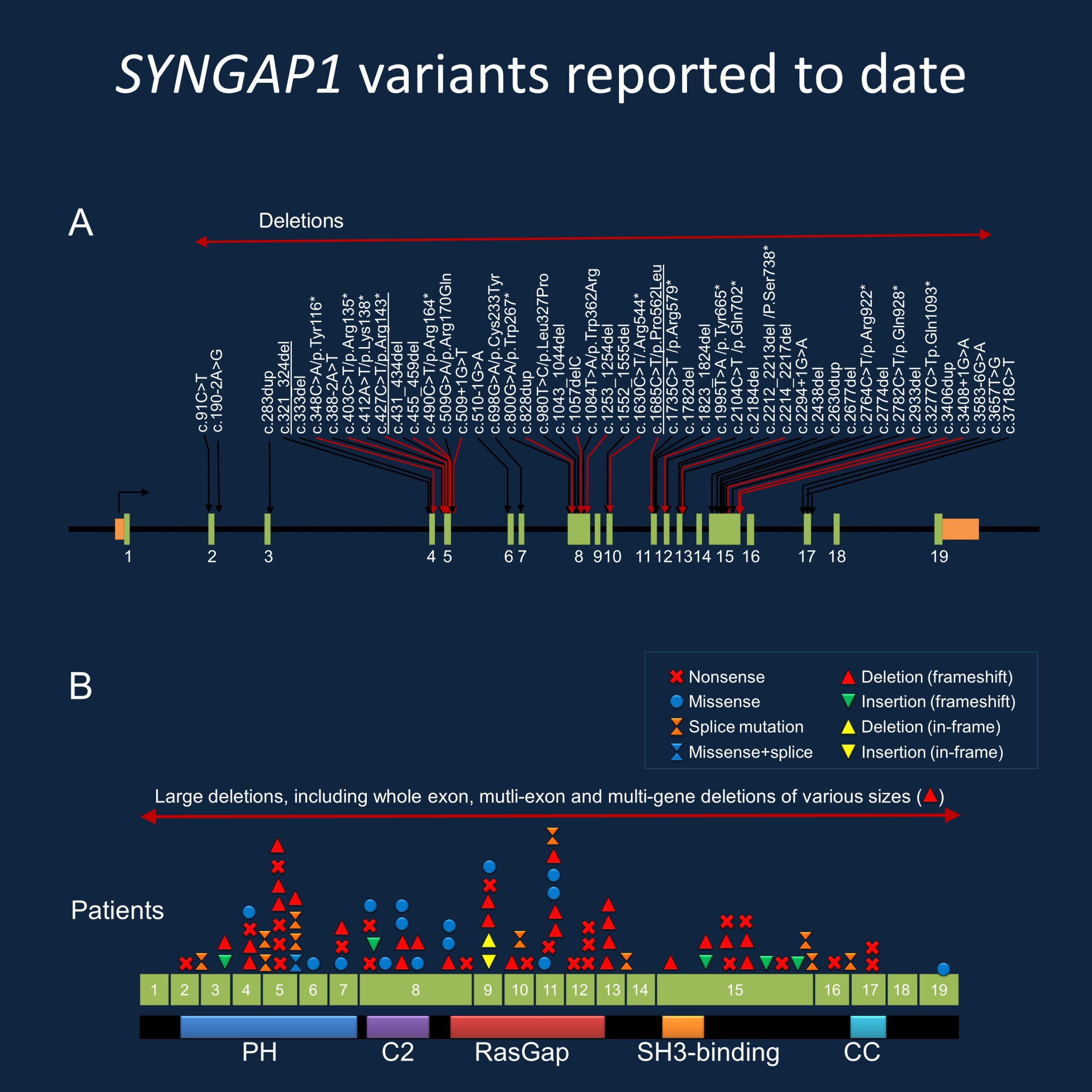
Figure 1. Spectrum of disease-causing variants in SYNGAP1. (A) Selection of variants reported in the literature since 2015, updated since our prior blog post. (B) Types of variants reported in the SYNGAP1 gene, demonstrating the wide range of genetic changes identified through diagnostic testing.
Phenotypes
Summary. SYNGAP1 is primarily associated with three main phenotypes, including non-syndromic intellectual disability, autism, and generalized epileptic encephalopathy. Over 100 affected individuals have been described in the literature to date, with many individuals having a combination of these features with varying severity.
Intellectual Disability/Developmental Delay. Almost all individuals with SYNGAP1-related developmental and epileptic encephalopathy (DEE) are diagnosed with intellectual disability (ID). Most individuals described have moderate to severe ID, while a minority of individuals have mild ID. There are rare individuals reported to have typical or nearly typical cognition. Global developmental delay is reported in the majority of individuals with expressive and receptive language development more severely affected than motor abilities. Developmental delays (DD) are noted to precede seizure onset in many individuals affected by both clinical features, and developmental plateauing or regression has been observed with seizure onset. Autism spectrum disorder (ASD) is diagnosed or suspected in over half of individuals.
Epilepsy. Generalized epilepsy is primarily associated with SYNGAP1-related DEE. Most, but not all, of affected individuals carry an epilepsy diagnosis. The average age of seizure onset is 2 years, but the age range spans infancy to adolescence. Individuals can experience multiple seizure types over their lifetime, including typical and atypical absences, atonic, and/or myoclonic seizures. Epilepsy with eyelid myoclonia with absences (EMA) and epilepsy with myoclonic-atonic seizures (MAE or Doose syndrome) can distinguish SYNGAP1-related epilepsy from other epileptic encephalopathies. Seizures with clinical and EEG photosensitivity have also been recently described. Less common seizure types include focal or infantile spasms. In addition, the more recent literature on SYNGAP1 has emphasized the relatively high rate of reflex seizures in individuals with SYNGAP1. Seizure severity does not seem to correlate with autism or the degree of cognitive impairment. To date, there is no known antiepileptic medication which is specific to pathogenic SYNGAP1 variants. Approximately half of individuals with epilepsy are treatment-resistant while others are well-controlled on medication.
Additional Emerging Features As genetic testing becomes both more widespread and accessible, and as characterization of SYNGAP1-related DEE advances, additional clinical features have emerged. Significant behavior problems are consistently reported in individuals with or without an ASD diagnosis, including oppositional and defiant behavior, aggression, self-injurious behavior, and sleeping difficulties. Many individuals affected by SYNGAP1 report feeding difficulties spanning poor feeding and oral aversion to gorging. A high pain threshold to adverse stimuli is also reported in a significant proportion of individuals. Hypotonia, ataxia and/or gait abnormalities are noted in slightly less than half of affected individuals.
Genotype
Genotype vs. phenotype. There is no known genotype-phenotype correlation in the SYNGAP1 gene. In a study by Vlaskamp and collaborators, a milder clinical phenotype of SYNGAP1-related DEE was more frequently associated with pathogenic or likely pathogenic variants in exons 1 to 4 compared to exons 5 to 19. However, variable phenotypes were still observed in individuals with the same variant, including siblings. In the study by Mignot and collaborators, there was some indication that patients with pathogenic variants in exons 4-5 are pharmacosensitive, while patients with variants in exons 8-15 tend to have intractable seizures. As the spectrum of SYNGAP1-related DEE is more fully elucidated, it will be interesting to see what genotype-phenotype correlations emerge.
Recurring variants. There are 15 recurrent variants within SYNGAP1 reported to date, although correlations between variants and clinical features remain elusive. These 15 known recurrent variants include: p.Lys114Serfs*20, p.Tyr116*, p.Lys138*, p.Arg143*, p.Arg164*, p.Arg170Gln, p.Lys277Glnfs*7, p.Lys418Argfs*54, p.Pro562Leu, p.Arg579*, p.Leu595Cysfs*55, p.Gln702*, p.Asn729Thrfs*31, p.Val1078Alafs*51, p.Gln1136Profs*17.
My patient has a mutation in SYNGAP1 – what does this mean? Assessing SYNGAP1 variants is difficult in many cases. This gene is included in many gene panels and clinicians may be faced with the problem to interpret milder or less frequent symptoms in probands correctly . For example, when looking at the evidence of SYNGAP1 variants being diagnostic, does the presence of nonspecific ID, generalized epilepsy, or only a diagnosis of ASD in the proband suggest that the variant is causative? Alternatively, as almost all known cases of SYNGAP1 arose de novo, does the inheritance of a germline variant from a parent equate to the variant not being diagnostic? SYNGAP1 is a large gene and with the advent of widespread genetic testing, both causative and rare population variants in this gene are identified. Here are three criteria that may help you interpret SYNGAP1 mutations in your patient.
1 – Variant. SYNGAP1 DEE is caused by pathogenic variants leading to haploinsufficiency. Numerous heterozygous loss-of-function variants have been reported, including nonsense, frameshift, and splice site variants, as well as microdeletions involving part or all of the SYNGAP1 gene. Pathogenic missense variants have also been observed. While there are 15 recurrent variants to date, the clinical presentations of individuals sharing the same genetic change are variable. However, penetrance is expected to be 100%. Therefore, either known recurrent variants or de novo loss-of-function variants in an individual with clinical features suspicious of SYNGAP1 may indicate a diagnosis. Functional data suggesting a loss of function for a particular variant may also suggest a possibility of SYNGAP1 DEE.
2 – Segregation. SYNGAP1-associated disorders are inherited in an autosomal dominant fashion and segregation data (testing parents and families) may help figure out whether a variant is pathogenic. Each child of an individual with a disease-causing variant has a 50% (1 in 2) of inheriting the disease-causing variant. Almost all reported variants with segregation analysis available have been de novo. At least one individual has been reported to inherit a pathogenic SYNGAP1 variant from a parent with somatic mosaicism. Although not yet reported, germline mosaicism of SYNGAP1 variants is possible, making the recurrence risk for families with one child with a SYNGAP1 variant higher than the general population risk.
3 – Phenotype. The phenotypic spectrum of SYNGAP1 is broad and continues to expand as more diagnoses are made and as research advances. While SYNGAP1 is primarily characterized by ID, DD/ASD, and generalized epilepsy, variable expressivity is observed even with those sharing the same pathogenic variant. Newly diagnosed individuals are expanding either end of the SYNGAP1 spectrum, and additional clinical features including focal seizures, behavioral problems and ataxias are arising. The spectrum of the phenotypes is broad and continuing to be described, and future studies will help us better understand the associated epilepsies and neurodevelopmental phenotypes.
Mechanism
Synapses. SYNGAP1 encodes for an essential protein of the post-synaptic density (PSD). The SYNGAP1 protein is a regulatory component of the postsynaptic density that is involved in neuronal plasticity. The effect of SYNGAP1 protein on the maturation of dendritic spines appears to inhibitory and reduced or absent SYNGAP1 protein may lead to enhanced maturation of dendritic spines. The SYNGAP1 protein is strategically placed in the postsynapse to be an interface between the NMDA receptors at the postsynaptic membrane and the signaling apparatus in the postsynapse that modulate the architecture of the dendritic spine. The underlying mechanism in SYNGAP1 encephalopathy is haploinsufficiency, i.e. the lack of two functional copies of the SYNGAP1 gene.
Cell/Animal models
Mice. Heterozygous Syngap1 knock-out mice exhibit deficits in hippocampal long-term potentiation (LTP), while re-expression of Syngap1 in adult mice rescues hippocampal LTP. This suggests that SYNGAP1 plays a crucial role in the regulation of synapse plasticity throughout the lifespan. Gou and collaborators have also illustrated that SynGAP expression in developing mouse neurons additionally regulates postnatal maturation of excitatory synapses.
iPSC. Llamosas and collaborators showed preliminary evidence that reducing SynGAP protein expression in induced pluripotent stem cell lines (hiPSC) resulted in enhanced dendritic morphogenesis, larger neuronal size and dendritic fields, increased excitatory synapses, and stronger excitatory activity expressed earlier in development compared to wild-type human iNeurons.
Community
Research studies. The scientific community is actively studying SYNGAP1 and its role in human disease. The SYNGAP1 Epilepsiome team is happy to facilitate if you have questions or a specific interest in this gene.
Advocacy organizations. The SYNGAP1 community is very active. Both the SynGAP Research Fund and the Bridge the Gap-SYNGAP Education and Research Foundation maintain websites about the most recent findings and research activities regarding SYNGAP1-related conditions. The SYNGAP1 Research maintains a list of studies you can access here.
Patient registry. The Bridge the Gap-SYNGAP Education and Research Foundation has partnered with several groups for their registries, including Simons Searchlight, Rare-X, and the Rare Epilepsy Network.