
CHD2. This is the Epilepsiome page for CHD2, a common cause of an early-onset epileptic encephalopathy. Exquisite environmental photosensitivity is the hallmark of CHD2.
Here are the most recent blog posts on CHD2
- CHD2 – this is what you need to know in 2015
- Flickering lights, endophenotypes, and EEG genetics – CHD2 in photosensitivity
- CHD2 myoclonic encephalopathy – delineating a novel disease
- CHD2 encephalopathy as a novel Dravet-like epilepsy syndrome
In a nutshell. Mutations in CHD2 are one of the more common causes of epileptic encephalopathy. From the time of our first blog post, CHD2 has also been associated with a wide spectrum of phenotypes, including self-limiting or drug-responsive epilepsy and neurodevelopmental disorders without seizures. The most commonly reported CHD2 phenotype has Dravet features (early onset, some have febrile seizures, myoclonic seizures), Lennox Gastaut features (multiple seizure types including tonic seizures) and Doose features (photosensitivity, myoclonic-astatic seizures). However, it is the exquisite environmental photosensitivity that is the hallmark of the epilepsy associated with CHD2.
| Phenotypes | Genetics | Mechanism & Function |
| The Clinical Perspective | Community | Resources & References |
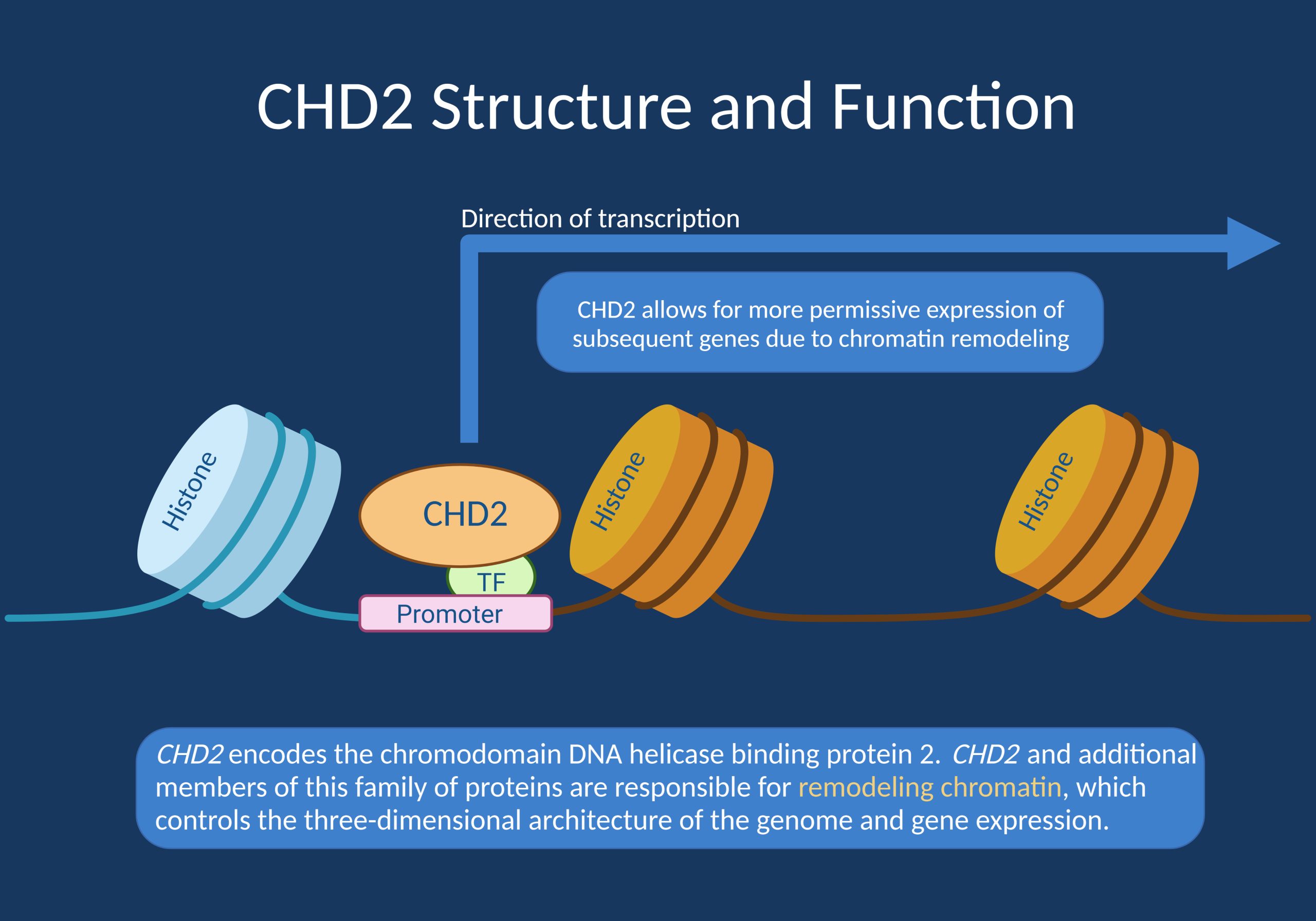
Figure. Cartoon depicting the function of CHD2. In brief, CHD2 stands for chromodomain DNA helicase binding protein 2, which is involved in chromatin remodeling. DNA is typically tightly packed around histones and therefore inaccessible to transcription. CHD2 is one of the many proteins involved in making DNA accessible for transcription and gene expression [created with BioRender].
Phenotypes
Epilepsy. Over 90% of individuals with CHD2 will develop seizures, with over 75% of seizures being generalized versus focal. Many patients with CHD2 mutations have an onset of seizures between age 6 months and 4 years with a mean onset of 2 years 6 months. However, there are reports of adult-onset epilepsy as well. More than half of affected individuals have multiple seizure types, with a predominance of myoclonic-atonic, myoclonic, and absence seizures as well as epilepsy with myoclonia. Other seizure types include drop attacks and rapid onset of multiple seizure types associated with generalized spike-wave on EEG. Seizures may be fever-sensitive. A seizure type observed in several patients has been called ‘atonic-myoclonic-absence seizure,’ and is characterized by a progressive seizure pattern with an abrupt head nod and atonia followed by a myoclonic absence phase and progression to a ‘ratchet-like’ tonic abduction of the upper limbs. Clinical photosensitivity, with seizures triggered by photostimulation such as a flickering light or television is a common feature and may be a distinguishing feature for such early-onset epilepsies. CHD2 mutations are also seen in common epilepsies associated with photosensitivity and in 3 of 36 patients with the most photosensitive epilepsy syndrome, eyelid myoclonia with absences. About 10% of individuals with CHD2 will have epilepsy with eyelid myoclonia.
Developmental delay and autism. Patients with CHD2 mutations may also have developmental delays that are often present prior to onset of seizures. About 82% have intellectual disability, with about 30% mild, 25% moderate, and 15% severe-profound. Autism spectrum disorder or autistic features are present in over half of individuals with CHD2, and behavioral issues are present in over 40%.
Other features. Patients with microdeletions of CHD2 and nearby genes can also have additional features, including truncal obesity, scoliosis, neonatal hypotonia and facial dysmorphism. However, it is unclear whether these findings are also relevant to patients with CHD2 sequence variants.
Phenotypes can vary, even within a single family with the same variant.
Genetics
Mutation Spectrum (Type and Location). Thus far, the majority of clear disease-causing mutations have been truncating (frameshift or nonsense) variants. Microdeletions (including CHD2 and surrounding genes) and missense and splice site variants have also been reported. Variants have been reported throughout the CHD2 gene and are not restricted to a single domain or exon. Most missense variants occur in functional domains, particularly the two helicase domains of CHD2.
Genotype-Phenotype Correlation. No clear genotype-phenotype correlations have been reported.
General Considerations for Variant interpretation. When reviewing a genetic variant to determine its significance for a given patient, it is important to weigh multiple pieces of evidence:
Gene level interpretation. First, it is important to establish the strength of the evidence showing that the gene is associated with epilepsy. Some genes may only have one variant reported in a single individual with epilepsy, while other genes may have multiple variants reported in many large families with an autosomal dominant pattern of epilepsy.
Variant level interpretation. When reviewing the significance of a variant, it is important to consider the impact on the gene and the presence of the variant in previously described patient and control populations. Many clinical genetic testing laboratories classify genetic variants into different categories, ranging from benign to pathogenic. Variants that are common in control populations and would not be predicted to have a major impact on the gene/protein are generally classified as benign. Variants are more likely to be classified as pathogenic if the variants are rare or not present in the control population, reported in multiple individuals or families with disease, and likely to have a higher impact on the gene/protein based on the type of mutation or functional studies. Variants with uncertain or limited available evidence may be classified as variants of uncertain significance (VUS), indicating that further information is required in order for the variant to be further defined. In some cases, testing additional family members can be helpful, as it allows the lab to determine whether or not the variant was inherited (versus de novo) and how the variant segregates with disease in the family. Sometimes further classification of a VUS requires waiting for the identification of additional patients or families with similar or nearby variants.
Inheritance, Penetrance & Prevalence. Many, if not all, clearly disease-causing CHD2 mutations have been de novo. Some variants have been reported to be inherited from unaffected parents, raising the possibility of incomplete penetrance or variable expressivity, but no cases of definite autosomal dominant inheritance or inheritance from an affected parent have been reported. The prevalence of CHD2-associated disorders is unknown.
Mechanism & Function
Haploinsufficiency. Most of the well-established disease-causing mutations that have been described are heterozygous truncating mutations or whole gene deletions, suggesting that haploinsufficency is the mechanism of disease. The disease mechanism of missense mutations is unknown.
Chromatin remodeling and cellular control. CHD2 encodes the chromodomain DNA helicase binding protein 2. CHD2 and additional members of this family of proteins are responsible for remodeling chromatin, which controls the three-dimensional architecture of the genome and gene expression. It also plays a crucial role in neurogenesis for both cortical excitatory and inhibitory neurons, as well as controlling the proliferation and differentiation of various cell types. Finally, CHD2 plays a role in DNA repair via its role in remodeling of chromatin. CHD2 is ubiquitously expressed. The mechanism of the epilepsy phenotype in individuals with harmful variants in CHD2 may be an imbalance of excitatory and inhibitory inputs, though more work is needed to clarify the specific mechanism of action.
Cell/Animal models.
Mice. According to Kulkarni et al (2008), mice with complete disruption of Chd2 (homozygotes) show embryonic/perinatal lethality, while mice with partial disruption of Chd2 (heterozygotes) have pronounced lordosis, kyphosis, absent or hypoplastic subcutaneous fatty tissues, and growth retardation.
Zebrafish. Zebrafish with partial loss of Chd2 function (i.e. Chd2 partially knocked down via morpholino) have abnormal movements, epileptiform discharges, and photosensitivity, as well as multiple developmental abnormalities not reported in humans with CHD2 sequence mutations, including body curvature, microcephaly, and stunted growth.
The Clinical Perspective
Recurrence Risk. Most disease-causing variants identified thus far are de novo or sporadic. A few families have been reported, mostly with affected parents. Germline mosaicism of CHD2 mutations is possible, making the recurrence risk for families with one child with a CHD2 mutation higher than the general population risk.
Therapy. Thus far, there is no specific recommended treatment regimen for patients with CHD2 mutations.
Community
Family Foundation. The Coalition to Cure CHD2 is a very active group involved in education, support, and research. They have many resources on their site and will have their inaugural family and scientific conference in June 2023.
Family page. The CHD2 patient community maintains an active Facebook page that researchers and families can join. The Facebook page is managed by Melanie Brazil, who will introduce you to the community if you would like to join.
Research studies. The scientific community is currently actively studying CHD2 and its role in human disease. The CHD2 Epilepsiome team is happy to facilitate if you have questions or a specific interest in this gene.
Resources & References
Websites
GeneReviews
Carvill G, Helbig I, Mefford H. 2015. CHD2-Related Neurodevelopmental Disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. PMID: 26677509
References
Carvill et al. 2013 Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45(7):825-30. PMID: 23708187
Chenier J et al. 2014. CHD2 haploinsufficiency is associated with developmental delay, intellectual disability, epilepsy and neurobehavioural problems. Neurodev Disord 2014;6(1):9. PMID: 24834135
De Maria et al. 2021. Expanding the genetic and phenotypic spectrum of CHD2-related disease: From early neurodevelopmental disorders to adult-onset epilepsy. AJMG Part A 188 (2), 522-533. PMID: 34713950
Galizia et al. 2015. CHD2 variants are a risk factor for photosensitivity in epilepsy. Brain 138:1198-207. PMID: 25783594
Kulkarni et al. 2008. Disruption of chromodomain helicase DNA binding protein 2 (CHD2) causes scoliosis. Amer J Med Genet Part A 146A:1117–27. PMID: 18386809
Lamar and Carvill 2018. Chromatin Remodeling Proteins in Epilepsy: Lessons From CHD2-Associated Epilepsy. Front. Mol. Neurosci Volume 11 – 2018. PMID: 29962935
Liu et al. 2015. Human CHD2 is a chromatin assembly ATPase regulated by its chromo- and DNA-binding domains. J Biol Chem 290(1):25-34.
Lund et al. 2014. CHD2 mutations in Lennox-Gastaut syndrome. Epilepsy Behav 33:18-21. PMID: 24614520
Suls et al. 2013. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 93(5):967-75. PMID: 24207121
Thomas et al. 2015. CHD2 myoclonic encephalopathy is frequently associated with self-induced seizures. Neurology 84(9):951-8. PMID: 25672921
Trivisano et al. 2015. CHD2 mutations are a rare cause of generalized epilepsy with myoclonic-atonic seizures. Epilepsy Behav 51:53-6. PMID: 26262932