
GABRA1. This is the epilepsiome page on GABRA1. A gene encompassing a wide spectrum of phenotypes; from JME to EE. Particularly interesting is the apparent photosensitivity in these patients.
Here are the most recent blog posts on GABRA1
- The two faces of GABRA1 – from familial epilepsy to epileptic encephalopathy
- Zeroing in on FAME – the dilemma of familial epilepsies
- GABRA1 and STXBP1 as novel genes for Dravet Syndrome
- Papers of the week – GABRA1 and STXBP1 in Dravet, gene therapy & synonymous mutations in cancers
In a nutshell. GABRA1 encodes the α-subunit of the GABA-receptor, GABA being the most widespread inhibitory neurotransmitter of the human brain. GABRA1 was first described in patients with mild genetic generalized epilepsy (JME), but since the phenotypic spectrum has expanded considerably to encompass more severe epilepsy forms, such as Dravet Syndrome.
| Phenotypes | Genetics | Mechanism & Function |
| The Clinical Perspective | Community | Resources & References |
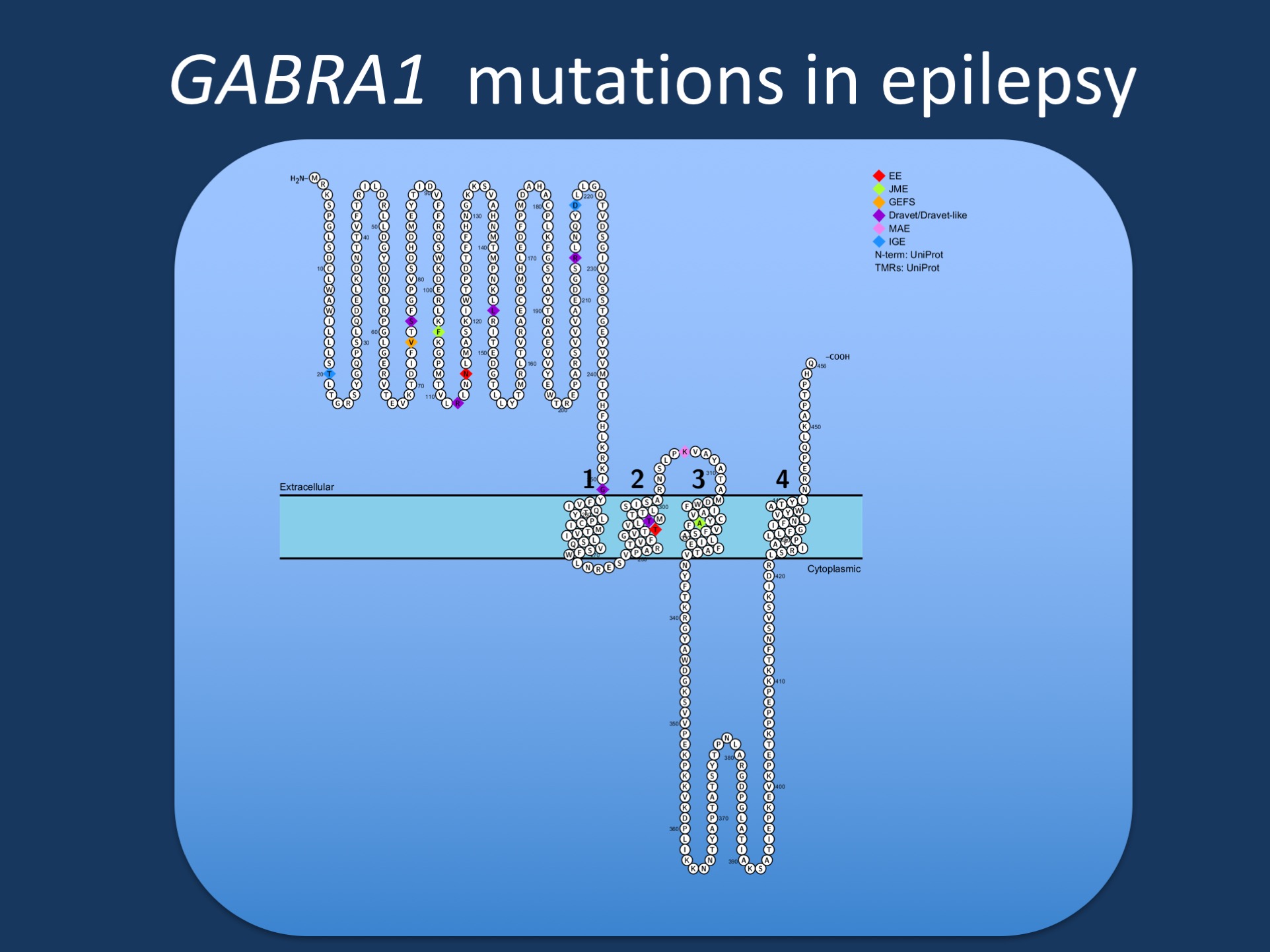
Figure. Pathogenic variants in GABRA1 in various phenotypes reported by Johannsen and collaborators. GABRA1 was initially identified in patients with Juvenile Myoclonic Epilepsy (JME), but most reported patients to date have more severe epilepsies such as epileptic encephalopathies (EE), Juvenile Myoclonic Epilepsy (JME), Dravet Syndrome, or Myoclonic Astatic Epilepsy (MAE). Few patients have milder phenotypes such as Genetic/Generalized Epilepsy with Febrile Seizures Plus (GEFS+).
Phenotypes
Summary: A wide spectrum of phenotypes is observed in patients with GABRA1 mutations, and include JME, IGE, Dravet syndrome, EE, as well as patients with just a few seizures, but otherwise normal intellect and development, and apparent seizure freedom. Shared features in GABRA1 patients include EEGs with generalized spike-wave discharges, half with a photoparoxysmal response. Most common seizure types are tonic-clonic and myoclonic seizures.
JME: One of the first phenotypes to be associated with GABRA1. Seizure types include myoclonic seizures, generalized tonic clonic seizures and absences. All the known variants with this phenotype have been inherited from an equal or milder affected parent. Intellectually the patients are normal. EEG can display generalized spike-wave discharges.
IGE: Only a few patients so far have been described with a GABRA1 variant and an IGE phenotype. The variant often segregates with the phenotype. Seizures include generalized tonic clonic seizures and febrile/afebrile tonic seizures. Intellect is normal. EEG displays generalized spike-wave discharges.
Dravet Syndrome: A large proportion of patients with GABRA1 have classical Dravet Syndrome, or a Dravet-like phenotype. Consistent with the classification, these patients have febrile seizures with onset between 6 and 12 months. Further seizure types develop as the patient grows older.
Epileptic Encephalopathy: The EE group encompasses a wide variety of more or less severely affected. Common for these patients is a wide variety seizure types, usually with a debut before the age of 1. All the patients suffer from intellectual disability, and have difficult to treat seizures.
Genetics
Genotype vs. phenotype: Currently, we are aware of approximately 28 different patients with almost as many GABRA1 variants. However, no clear genotype-phenotype correlation has emerged from these cases. Genotype has not been able to explain the clinical heterogeneity. The majority of the identified GABRA1 mutations are missense and arise de novo whereas only about one third follows an AD inheritance. Interestingly, there seems to be phenotypic concordance within the families with segregation of mutation and symptoms, which is different from the SCN1A mutations, in which phenotype can differ within the same family.
Recurrent mutations: Six mutations were observed to be recurrent so far; Ser76Arg, Arg112Gln, Arg214His, Gly251Ser/Asp, Thr289Pro/Arg and Lys306Thr.
Ser76Arg: Both patients shared a similar phenotype characterized by hemiclonic/hemiclonic-tonic seizures at onset, subsequent intractable myoclonic seizures, and severe developmental delay with autistic features.
Arg112Gln: mutation has been detected in patients with severe EE phenotype, but also in a girl with only a few seizures so far and normal psychomotor development.
Mechanism & Function
Mechanism. Functional studies indicate that mutations cause disease via haploinsufficiency and/or through a dominant negative effect on wild-type partnering subunits. The suggested pathomechanism of previously identified mutations is reduced protein stability and surface expression of GABAA receptors comprising mutant subunits, as well as decreased sensitivity to GABA, resulting in a loss-of-function and subsequent reduction of inhibitory inputs in neurons and therefore increased excitability. A recent study confirmed the loss-of-function mechanism in several variants. The majority of identified missense GABRA1 mutations localizes at the N-terminus of the α1-subunit, which contains the putative signaling peptide and is responsible for binding GABA and its agonists. Other affected regions are the transmembrane segments M1, the pore-forming M2 and M3, as well as the extracellular M2-M3 linker involved in the coupling of agonist binding with gating. Thus, the described GABRA1 mutations affect regions critical for the proper function of GABAA receptors.
The Clinical Perspective
My patient has a mutation in GABRA1 – what does this mean? Assessing GABRA1 variants is difficult in many cases. This gene is included in many gene panels and clinicians may be faced with the problem of interpreting milder symptoms in transmitting carriers correctly. Unfortunately, so far, no ideal way of separating the milder phenotypes from the more severe exists as of yet. A positive family history will point towards the more mild phenotypes. Genetic background or modifier genes might contribute to the clinical diversity.
Community
There is currently no research community or patient organization for patients carrying GABRA1 variants.
Resources & References
Websites