
SLC6A1. This is the Epilepsiome page for SLC6A1, a gene for Doose syndrome, or myoclonic atonic epilepsy (MAE).
Here are the most recent blog posts that mention SLC6A1:
- SLC6A1 – a generalized epilepsy phenotype emerging
- Identifying the Doose gene – SLC6A1 mutations in Myoclonic Astatic Epilepsy
In a nutshell. SLC6A1 codes for GAT-1, a GABA transporter that removes GABA from the synaptic cleft. Loss of function of SLC6A1 is a common cause of myoclonic atonic epilepsy (MAE), also known as Doose syndrome. Features include mild to moderate intellectual disability and myoclonic, absence, and atonic seizures.
Phenotypes | Genetics | Mechanism | Community
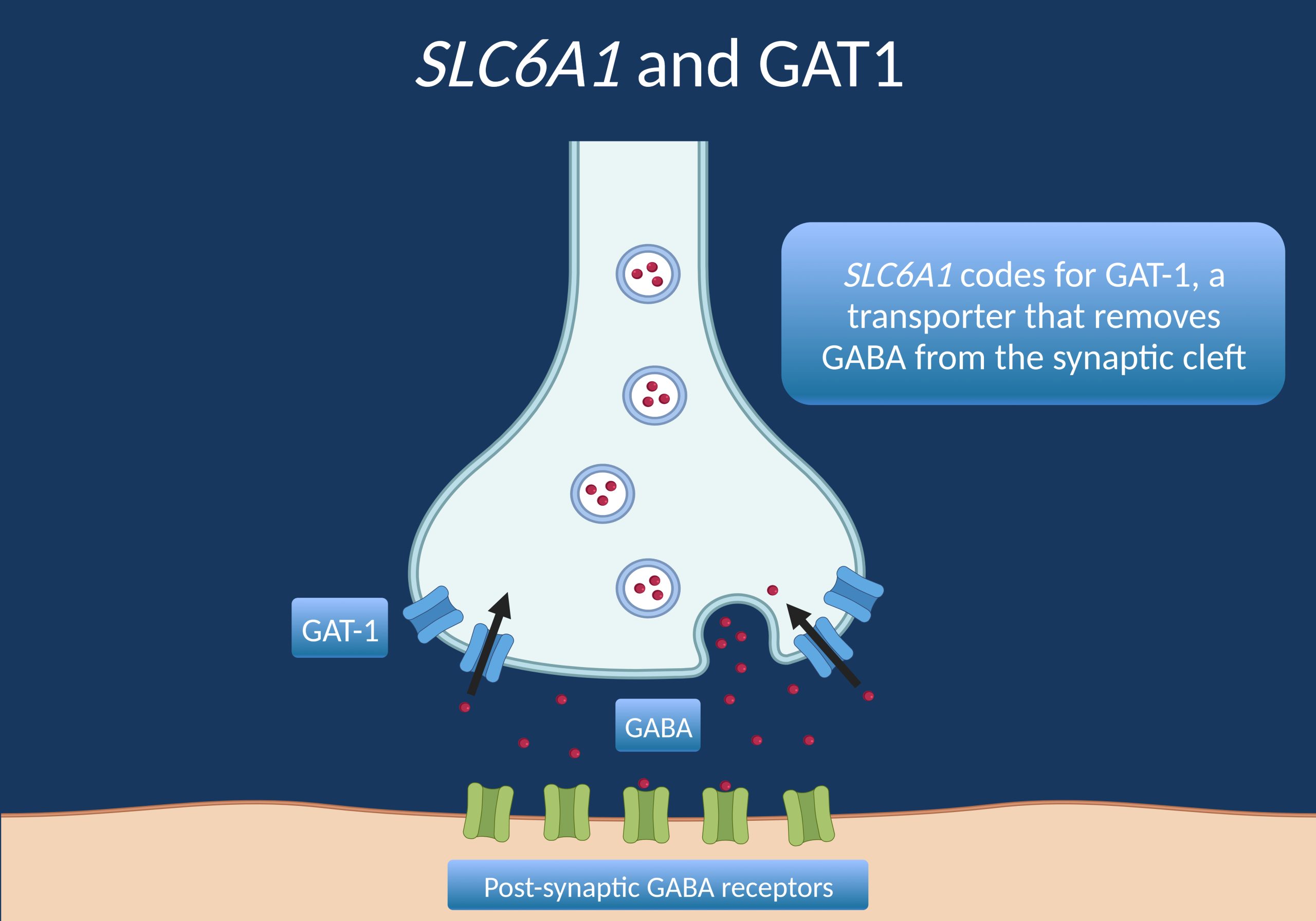
Figure 1. Diagram of the synaptic cleft where GAT-1, the transporter protein made by the SLC6A1 gene, reuptakes GABA into the pre-synaptic neuron.
Phenotypes
Summary. Doose (“dohs-ah”) syndrome was identified in the 1970s as a epilepsy syndrome involving myoclonic-astatic or myoclonic-atonic seizures, and was eventually renamed as Myoclonic Atonic Epilepsy (MAE). SLC6A1 was the first gene discovered to cause MAE, and has remained one of the most common causes of it. The phenotype is generally limited to epilepsy and mild to moderate cognitive impairments. While the severity and range of specific features has widened over the years, the phenotype has remained relatively constrained compared to some other epilepsy genes.
Seizures. Myoclonic atonic epilepsy (MAE), also know as Doose syndrome, is one of the most common presentations in SLC6A1-related disorders. However, other seizure types can present as well. Goodspeed and colleagues (2020) reported on 116 individuals with SLC6A1, and found that 91% had seizures. Mean age of onset was 2.5 years, with a standard deviation of 1.5 years. The most common epilepsy syndrome, as expected, was early-onset epilepsy with myoclonic atonic seizures, which presented in 24% of individuals. Genetic generalized epilepsy was present in 23%. Focal epilepsy was also relatively common, presenting in nearly 10% of individuals. In terms of specific seizures types, common ones included absence (72%), atonic (44%) and myoclonic (28%). Kahen and colleagues (2021) similarly found that 82% of their cohort of 28 individuals had seizures. Of those that had seizures, the most common types were absence (87%), atonic (57%), and myoclonic (52%).
Cognition. Goodspeed and colleagues (2020) found that most individuals with SLC6A1 present with developmental delay, especially after the onset of seizures (60% before seizure onset and 84% after seizure onset). Delays were mostly in the mild to moderate range. In contrast, Kahen and colleagues (2021) found that all individuals in their cohort of 28 did indeed have either intellectual disability or developmental delay. Autistic traits were present in 23% of the Goodspeed and colleagues cohort, while autism spectrum disorder was present in 29% of the Kahen and colleagues cohort.
Other neurological features. Hypotonia is common (60% per Goodspeed and colleagues, 2023) and improves with age. Movement disorders (primarily ataxia and intermittent tremor) are also relatively common, occurring in about 40% of individuals with an SLC6A1-related disorder. Finally, ADHD, sleep disturbances, and behavioral issues like aggression are also reported.
Genetics
Genotype vs. phenotype. The mechanism of SLC6A1-related disorders is expected to be loss of function or haploinsufficiency. No specific genotype-phenotype correlations exist. Currently, there are no well-established gain of function effects in SLC6A1-related disease.
Segregation. SLC6A1-related disorders are inherited in an autosomal dominant pattern. Most cases are de novo (~81%), though a relatively high proportion of cases (~19%) are inherited (Godspeed et al, 2020).
My patient has a mutation in SLC6A1– what does this mean? Assessing SLC6A1 variants is difficult in many cases. This gene is included in many gene panels and clinicians may be faced with the problem to interpret milder symptoms in transmitting carriers correctly. For example, when looking at the segregation of SLC6A1 variants, does the presence of seizures in a parent of child with MAE suggest that the variant is causative? With the advent of widespread genetic testing, both causative and rare population variants in this gene are identified. Here are three criteria that may help you interpret SLC6A1 mutations in your patient.
1 – Variant. Clear loss of function variants, such as whole gene deletions or truncating variants leading to nonsense-mediated decay, can be assumed to be likely pathogenic. In addition, many missense variants have been well-established as causing loss of function and leading to an SLC6A1-related neurodevelopmental disorder.
2 – Segregation. SLC6A1-associated disorders are inherited in a dominant fashion and segregation data (testing parents and families) may help figure out whether a variant is pathogenic. Severe, early onset childhood phenotypes in otherwise unaffected families may be de novo. Alternatively, the variant can be inherited from a parent with a milder phenotype. Given that the phenotype in SLC6A1-related disorders can be mild, careful collection of family history can be helpful in identifying cases where the disorder is inherited.
3 – Phenotype. The phenotypic spectrum of SLC6A1 is somewhat large and the question may arise – which phenotype would be considered incompatible with SLC6A1 as the underlying gene, suggesting that an identified variant may be considered non-causal based on the phenotype alone? Based on our current understanding of SLC6A1, there are a few seizure types (myoclonic, atonic, and absence) that are highly suggestive of an SLC6A1-related disorder. However, other seizures types including focal seizures have been seen. In addition, the cognitive profile tends to be mildly to moderately impaired, with few unaffected individuals or profoundly affected individuals. Future studies will help to elucidate the full phenotypic spectrum of SLC6A1-related disorders, as well as which presentations fall outside the possible spectrum.
Mechanism
GABA reuptake. SLC6A1 codes for GAT-1, a GABA transporter. GAT-1 removes GABA from the synaptic cleft, which limits the effect of GABA on the post-synaptic neuron and makes it available for future release in the pre-synaptic neuron. Loss of function or haploinsufficiency is the expected molecular mechanism. Pathogenic missense variants in the gene have been shown to impair protein trafficking, as well as impairing GABA uptake (Cai et al., 2019). The precise mechanism by which impaired GABA reuptake leads to seizures is not yet understood.
Community
SLC6A1 Connect is a very active family foundation, and several studies to elucidate natural history and work towards targeted therapies. SLC6A1 is also part of Simons Searchlight, which has published on their cohort of individuals.
References
Goodspeed K, Pérez-Palma E, Iqbal S, Cooper D, Scimemi A, Johannesen KM, Stefanski A, Demarest S, Helbig KL, Kang J, Shaffo FC, Prentice B, Brownstein CA, Lim B, Helbig I, De Los Reyes E, McKnight D, Crunelli V, Campbell AJ, Møller RS, Freed A, Lal D. Current knowledge of SLC6A1-related neurodevelopmental disorders. Brain Commun. 2020;2:fcaa170.
Goodspeed K, Mosca LR, Weitzel NC, Horning K, Simon EW, Pfalzer AC, Xia M, Langer K, Freed A, Bone M, Picone M, Bichell TJV. A draft conceptual model of SLC6A1 neurodevelopmental disorder. Front Neurosci. 2023 Jan 19;16:1026065. doi: 10.3389/fnins.2022.1026065.
Johannesen KM, Gardella E, Linnankivi T, Courage C, de Saint Martin A, Lehesjoki AE, Mignot C, Afenjar A, Lesca G, Abi-Warde MT, Chelly J, Piton A, Merritt JL 2nd, Rodan LH, Tan WH, Bird LM, Nespeca M, Gleeson JG, Yoo Y, Choi M, Chae JH, Czapansky-Beilman D, Reichert SC, Pendziwiat M, Verhoeven JS, Schelhaas HJ, Devinsky O, Christensen J, Specchio N, Trivisano M, Weber YG, Nava C, Keren B, Doummar D, Schaefer E, Hopkins S, Dubbs H, Shaw JE, Pisani L, Myers CT, Tang S, Tang S, Pal DK, Millichap JJ, Carvill GL, Helbig KL, Mecarelli O, Striano P, Helbig I, Rubboli G, Mefford HC, Møller RS. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia. 2018;59:389–402.
Kahen A, Kavus H, Geltzeiler A, Kentros C, Taylor C, Brooks E, Green Snyder L, Chung W. Neurodevelopmental phenotypes associated with pathogenic variants in SLC6A1. J Med Genet. 2022 Jun;59(6):536-543. doi: 10.1136/jmedgenet-2021-107694. Epub 2021 May 18.