
STX. We typically don’t blog about preprints, but we are making an exception this time given the upcoming STXBP1 Summit in Philadelphia on August 19-20. This is a post about one of our projects on STXBP1 that tries to understand the clinical presentation holistically, trying to find a way to capture the lived experience of families with STXBP1. In our current manuscript that will be presented at the STXBP1 Summit, we introduce our disease concept model for STXBP1. Disease concept models are formal frameworks that are increasingly required by regulatory agencies such as the FDA. Here is a brief overview what we find when we conduct formal interviews with families how a disease concept model helps us define phenotypes.
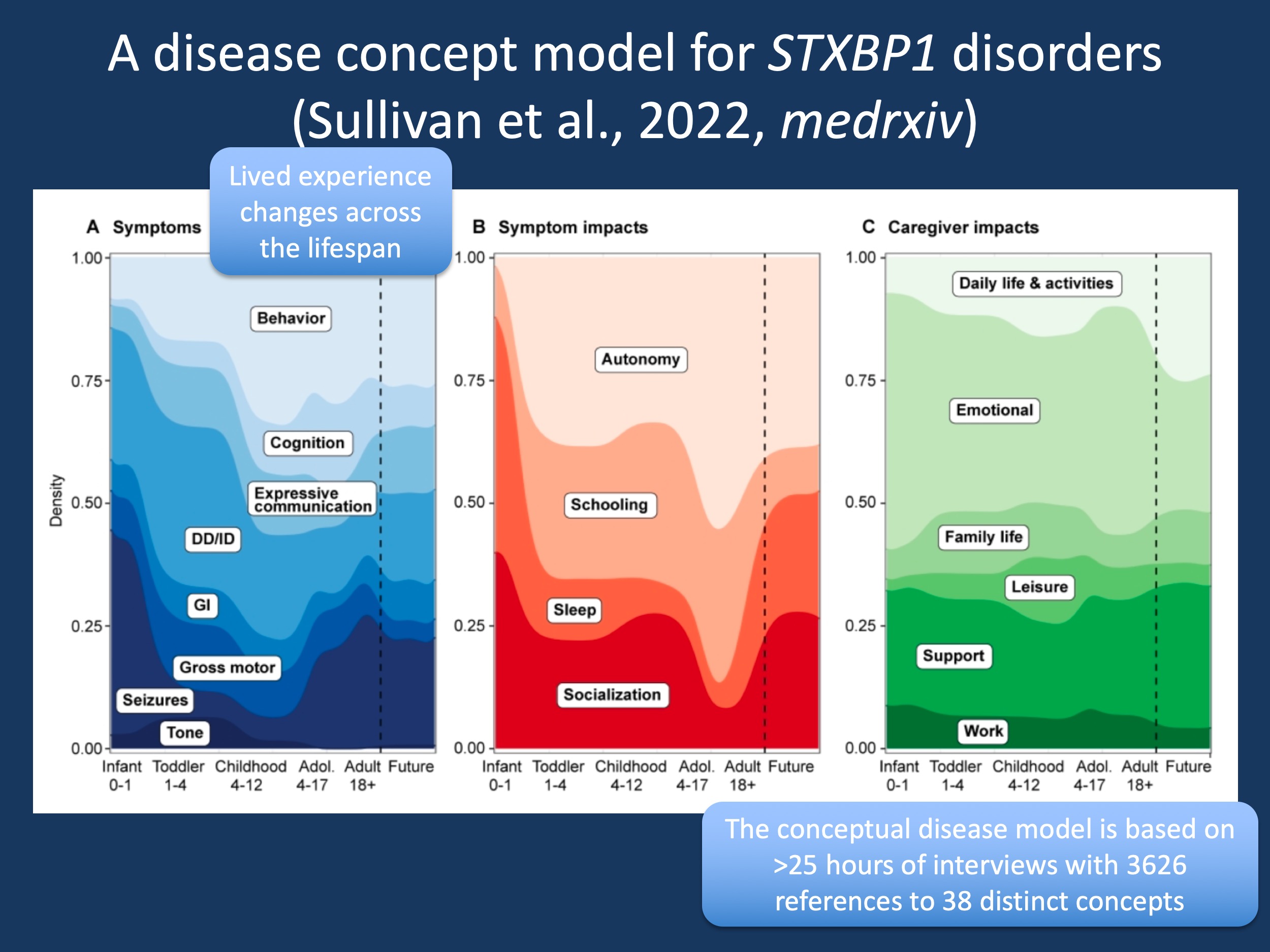
Figure 1. The changes in reported symptoms, symptom impacts and caregiver impacts across the lifespan in STXBP1-related disorders. While seizures predominate in infancy, concern about behavior and cognition are more prominent during childhood. When looking at symptom impact, autonomy is a critical feature beyond infancy. Categories of caregiver impact are relatively constant across the lifespan with emotional impact and support representing the two most common categories.
Disease concept. Our work on the STXBP1 disease concept model stems from Katie Rose Sullivan’s Genetic Counseling Master’s thesis. One aspect that I truly appreciate working with Genetic Counseling students is the fact that our team is constantly challenged to expand our horizon with regards to projects that we’re taking on. Katie Crawford’s thesis led to our publication on SCN2A, the first gene to be completely “digitized”, Stacey Cohen’s thesis introduced the concept of “days minimally impacted by seizures” to align Quality of Life with measures of seizures impact. Now, Katie Rose’s thesis pushed us into the domain of qualitative research, motivated by the seminal work in the field by Terry Jo Bichell on Angelman Syndrome. I have always approached qualitative research by pointing out that I’m not a qualitative researcher – I’m here to learn. And here is what our work on the STXBP1 disease concept model taught me.
How does this fit in. We will introduce the disease concept model (DCM) in detail during our upcoming meeting, so I wanted to use this blog post to explain how this fits into the larger picture on how we think about STX. Admittedly, qualitative research and patient interviews are not our forte, we are a bioinformatic, EMR lab of self-described phenotypic atomists. However, there is a link. It has to do with how we measure aspects of a disease and how we define the “search space”. In brief, DCMs answer the elementary question on what we are actually concerned with, they define the entirety of the symptoms that go along with a given condition. Given the broad range of phenotypes in STXBP1-related disorders, this is where we started.
What we did. In brief, we conducted patient interviews using an interview guide with 36 open questions, resulting in 25 hours of interview time with 3626 references to 38 concepts and an intercoder reliability of 88.2%. The interview guide was based on a preliminary DCM with features retrieved from the literature. We interviewed 19 caregivers and seven healthcare providers and ensured diversity in our study population with regards to age, race, sex, and STX clinical features. This was essential for our DCM to be generalizable to the ‘average’ STX family; accounting for the stories of differing clinical presentations (seizures history, ambulation, and expressive communication) as well as family structure (same sex couples, single parents, households that speak English as a second or third language). Healthcare providers consisted of physicians, genetic counselors, physical therapists, occupational therapists, and educators; mirroring the clinical and therapeutic team assembled for a newly diagnosed individual with STXBP1-related disorder. We transcribed each interview and coded concepts using the NVivo software, a HIPAA-compliant qualitative data analysis computer software. Coding was categorized in the following domains: symptoms, symptom impacts, and caregiver impacts. I was initially skeptical that this method would work, but once Katie Rose and our team got started, I was truly impressed by the scientific rigor of this approach. My takeaway is that qualitative research is not fuzzy or soft – it is a different way of doing science that has its own rules. And it adds to our general concept on how we think about developmental disorders, especially for a lab that is used to hard phenotype science.
What we found. Developmental delay, behavior, and seizures were the most common symptoms reported by caregivers, reflecting the burden of STXBP1-related disorders. Interestingly, we found frequency references to gastrointestinal and respiratory symptoms as well as pain, features that had not been reported previously. These results emphasize the purpose of a DCM – we can identify symptoms that may not be captured sufficiently given the compartmentalization of medical care. I am absolutely guilty of this – as a child neurologist, I am very likely one of the reasons that gastrointestinal and respiratory features are not as prominent as neurological features. We typically consider these issues secondary to the neurological features – however, when understanding the lived experience, there are no secondary features, but only features that define a family’s perception of a given condition.
Beyond symptoms. Symptoms were only one of the categories we looked at. We also examined symptom impact and caregiver impact, concepts that typically do not show up in patient letters and medical notes. We identified autonomy, socialization, and schooling as the main symptom impacts. Emotional impact, support, and impact on daily life and activities were the main caregiver impacts. Looking at these three domains, symptoms, symptom impact, and caregiver impact, allowed us to capture the entirety of the lived experience of STXBP1-related disorders. It is about casting a wide net and describing a condition in its entirety – this is what DCMs are meant to do.
From DCM to outcomes. By outlining the overall landscape across symptoms, symptom impact, and caregiver impact, DCMs provide a framework that will allow us to capture specific outcome measures. This is what a disease concept model does – it defines the domains that we need outcome measures for. For example, as you can see in Figure 1, seizures are only one of many features that drive the lived experience of STX. In reality, seizures merely intersect with behavioral, developmental, and communication symptoms and it is this amalgam that is much more important to families than any of these features alone. Accordingly, conceptualizing STX as a genetic epilepsy is somewhat shortsighted – we need more comprehensive ways to capture the impact of STXBP1-related disorders. I made a reference to STXBP1 in our prior post on WDR45-related disorder, where we have faced similar issues. Seizures, while easily measurable in studies, do not represent the true impact of a condition. In contrast, developmental issues are the main features.
This is what the DCM tells us. In brief, we were surprised by the following findings. First, non-neurological issues are very important in STXBP1-related disorders, they drive a significant component of the symptoms reported by families. Second, symptoms are only one of three domains reported by families. We need better measurements to capture the impact of symptoms rather than symptoms themselves as well as the impact on families. Third, the lived experience changes across the lifespan. There is no single measurement that described STXBP1-related disorders at any point in time in its entirety – when understanding the lived experience of neurodevelopmental disorders such as STXBP1-related disorders, we need to be aware that we are faced with an ever-changing picture of symptoms, their impact, and how they affect caregivers and the individual with a neurodevelopmental disorder.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
