
Semantic similarity. The phenotype era in the epilepsies has now officially started. While it is possible for us to generate and analyze genetic data in the epilepsies at scale, phenotyping typically remains a manual, non-scalable task. This contrast has resulted in a significant imbalance where it is often easier to obtain genomic data than clinical data. However, it is often not the lack of clinical data that causes this problem, but our ability to handle it. Clinical data is often unstructured, incomplete and multi-dimensional, resulting in difficulties when trying to meaningfully analyze this information. Today, our publication on analyzing more than 31,000 phenotypic terms in 846 patient-parent trios with developmental and epileptic encephalopathies (DEE) appeared online. We developed a range of new concepts and techniques to analyze phenotypic information at scale, identified previously unknown patterns, and were bold enough to challenge the prevailing paradigms on how statistical evidence for disease causation is generated.

Figure 1. A re-introduction to SCN1A-related epilepsies. What patterns do we see when we put together all the clinical information in EuroEPINOMICS, Epi4K, and other epilepsy studies and analyze data for patterns that are linked to de novo variants in SCN1A? The clinical features are displayed as a “phenogram,” comparing the frequency in individuals with SCN1A de novo variants (y-axis) to the larger cohort of developmental and epileptic encephalopathies (x-axis), with red dots indicating terms that are significantly different. On the right, the phenotypes listed are shown as a “phenotree,” displaying the connection of phenotypic features associated with SCN1A. Phenograms and phenotrees are some of the tools we developed to visualize large-scale phenotype data in the developmental and epileptic encephalopathies to make it more intuitive. This data, based on >30K single HPO terms, reconstructs the clinical features of Dravet Syndrome even though none of the included individuals have been clinically diagnosed.
Phenotyping gap. Yes, for those of you who have heard me give presentations in the last two years, I will sound like a broken record. However, let me say this again: we have to overcome the what I call the “phenotyping gap” in genetic epilepsies: the relative lack of clinical data compared to the large amount of genomic data that we are constantly generating. Phenotypic information is incredibly valuable in the genetic epilepsies, but generating and analyzing clinical information is difficult, especially when we are trying to merge information across datasets. Understanding treatment responses and outcomes are the new frontier in epilepsy genetics, but how should we go about this? More than two years ago, our group decided to explore a different, complementary approach to the classical clinical and natural history studies. We tried to find new ways to analyze clinical data that was already available, but was not meaningful by itself. Enter the era of computational phenotyping – we have developed methods to make this data speak to us and reveal some of the secrets of the developmental and epileptic encephalopathies. However, let me start our story somewhat differently.
Rett Syndrome. It does not happen very often that we get the opportunity to start a publication with a sentence like, “In 1954, Dr. Andreas Rett, a pediatrician in Vienna, Austria, noticed two girls with unusual repetitive hand-washing motions in his waiting room.” In our publication in AJHG, we used the example of Rett Syndrome as a basic introduction to the way we typically define clinical syndromes. Clinical syndromes, such as Rett Syndrome, are recognized because of shared clinical features that stand out. The cognitive process that identifies uncommon features shared by two individuals as a new disease entity is typical intuition grounded in expert knowledge – it is usually an automatic process, a “System 1: Thinking Fast” activity, as Daniel Kahneman would say. However, it is not as mysterious as it sounds. If we find a way to “weigh” rare clinical features appropriately, it would theoretically be possible to have an algorithm perform such a disease recognition. However, in order to get there, we need two key ingredients: data and concepts.
Phenotype data. It may sound difficult to believe based on what I mentioned earlier, but phenotype data is not missing and it is not difficult to obtain. Even for rare genetic epilepsies, the total observational timespan at many medical centers amounts to hundreds, if not thousands, of patient years. Likewise, in large projects such as EuroEPINOMICS and EPGP/Epi4K, large phenotypic datasets have been accumulated and the cost of phenotyping an individual sometimes even exceeds today’s cost of genome sequencing. The main issue is using the existing information, which is often stored in unique data formats and may be incomplete or “sparse” (there are many terms, but each term is only assigned to very few individuals). The first step to make such data useful is a process called data harmonization. In our study, we mapped all existing data onto a common format, namely the Human Phenotype Ontology (HPO). The HPO has the advantage that it is not only a dictionary, but has a tree-like structure that tells us about relationships. An “absence seizure” in Individual A is related to a “myoclonic seizure” in Individual B as both represent “generalized-onset seizures.” The amount of clinical data to be mapped in the joint EPGP/Epi4K and EuroEPINOMICS dataset, supplemented with clinical data derived from our DFG Research Unit and local data, quickly got quite large – we mapped a total of 31,000 phenotypic terms in 846 individuals with existing trio exome data. And yes, trio exome analysis was the easy part…
New concepts. Having a harmonized dataset then leads to the next set of question: how can this data be analyzed? We explored several approaches. First, with our harmonized dataset, we can explore how certain clinical features relate to known genetic etiologies. In order to make this data more intuitive, we developed two new ways to visualize such information, phenograms and phenotrees (Figure 1). You can easily see from the figure that such a way of visualizing data tells you information that a simple table cannot convey. For example, for the 16 individuals with de novo variants in SCN1A, we were able to reconstruct a phenotype profile that reflects the clinical features of Dravet Syndrome. Different patterns can be seen for genetic epilepsies due to the de novo variants in STXBP1, KCNQ2, or SCN2A that we included in the main part of our publication (other genetic etiologies are shown in the Supplement). Overall, we found more than 800 nominally significant gene-phenotype (HPO) associations in the developmental and epileptic encephalopathies.
Phenotypic similarity. Associations between phenotypic features are very informative, but when you look at Figure 1, you see more than just a number of associated terms – you see a profile and pattern. We used a method called phenotypic similarity analysis to ask the question whether the aggregated phenotypic features for each gene are more similar than we would expect by chance. A little more than one year ago, we used this method to identify a recurrent de novo variant in AP2M1 as a novel cause for developmental and epileptic encephalopathies. In our current study, we now expand this method to the larger group of DEEs, including information from all the trio epilepsy studies that were performed over the last decade. We found that 11/41 genes with two or more de novo variants were significantly similar, including SCN1A, STXBP1, and KCNB1. If we didn’t know anything about these genes, our analysis would have picked them out as likely disease-causing based on phenotypic features alone, without any control data and within a group of relatively similar phenotypes.
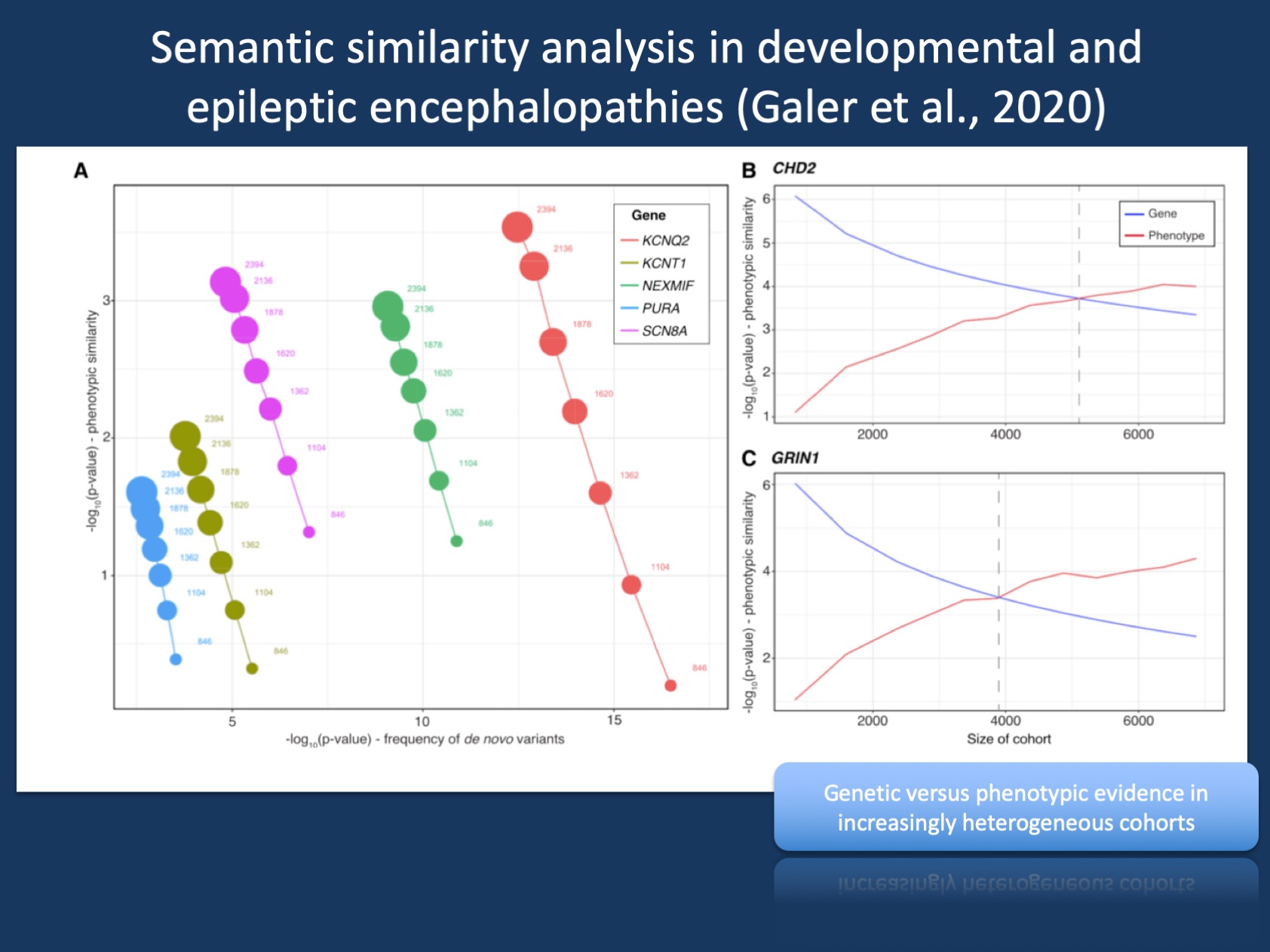
Figure 2. Phenotypic similarity analysis thrives on heterogeneity. Addition of controls results in increased phenotype-based significance and reduced genotype-based significance (A) On the basis of the initial cohort of 846 individuals with DEE, the subsequent addition of 1,548 population controls sequenced for de novo variants results in a steady increase in the statistical significance of gene-based phenotypic similarity. Inversely, statistical significance based on the frequency of observed de novo variants steadily decreases with the addition of controls. (B and C) With additional simulated controls, significance based on phenotypic similarity eventually exceeds significance based on frequency of de novo variants for CHD2 (B) and GRIN1 (C). The gray line indicates the critical cohort size when phenotypic significance becomes more significant than genotype-based significance.
Genetic vs. phenotypic evidence. Let me emphasize this last point again: without any control data and within a group of relatively similar phenotypes, our phenotype-based analysis lags behind the statistical power of genomic studies…for now. Virtually all genes in our cohort had far more extreme p-values when performing a conventional genetic analysis, assessing the likelihood of de novo variants in each gene. However, going back to the initial Epi4K publication and our subsequent EuroEPINOMICS/EPGP/Epi4K study that found DNM1, the type of analyses we had performed to push genes across the finish line has always struck us as somewhat odd. SCN1A, for example, is not a disease gene because the frequency of de novo variants in SCN1A is higher than expected by chance in a cohort of mixed DEEs. It is a disease gene as it represents a recognizable syndrome! We don’t need a genetic p-value to recognize that the neonatal-onset epilepsies in KCNQ2-related epilepsies or generalized epilepsies with features of Doose Syndrome in SLC6A1 represent disease entities. Our methods to provide statistical evidence based on phenotypes rather than frequency of de novo variants now allows us to re-think the way that we analyze combined genomic/phenotypic datasets. And this is not the end…
Phenotypes exceed genotypes. Admittedly, we’re not there yet. But what kind of analysis would you perform if you had to analyze an entire hospital or healthcare system with existing genomic and phenotypic data? In such a situation, every genetic etiology would be exceedingly rare and not significant. Phenotypic similarity analysis, however, thrives on heterogeneity (Figure 2). We added more than 1,500 control trios sequencing for de novo variants and the evidence for genes based on de novo variants went down. This was expected, as de novo variants in SCN1A will be diluted if you add thousands of controls. However, significance based on phenotypic similarity increased! If you add heterogeneous phenotypes, existing similarities between individuals with a common genetic cause are enhanced. We took this one step further and asked an almost heretical question: is there a point when phenotypic similarity analysis would be the better, more powerful analysis tool? The answer was yes – in heterogeneous cohorts upwards of 4,000 individuals, phenotypic similarity will likely beat assessments based on the frequency of de novo variants, which was traditionally one of the most powerful methods to generate statistical evidence so far.
What you need to know. Yes, this blog post is relatively long, but it was important to me to discuss our publication on HPO-based similarity in developmental and epileptic encephalopathy at length. Ever since the early days of EuroEPINOMICS, I was intrigued by computational methods to assess phenotypic relatedness (link) and to me, the publication of our final EuroEPINOMICS paper marks the beginning of a new era. We have shown that we can handle heterogeneous clinical data in conjunction with genomic data at scale and use these tools to generate new knowledge. The age of computational phenotyping has begun.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
