
KCNQ2. This is the Epilepsiome page on KCNQ2, an ion channel gene that encodes a subunit of a voltage-gated potassium channel. Known for years as a gene for a familial and self-limiting neonatal epilepsy syndrome, mutations are now also shown to be a frequent cause of neonatal epileptic encephalopathy. Here is what you need to know about KCNQ2.
Here are the most recent blog posts on KCNQ2
- KCNQ2 – this is what you need to know in 2015
- The OMIM epileptic encephalopathy genes – a 2014 review
- The top three publications in epilepsy genetics 25 years ago
- Mutation intolerance – why some genes withstand mutations and others don’t
Phenotypes | Genetics | Mechanism | Community
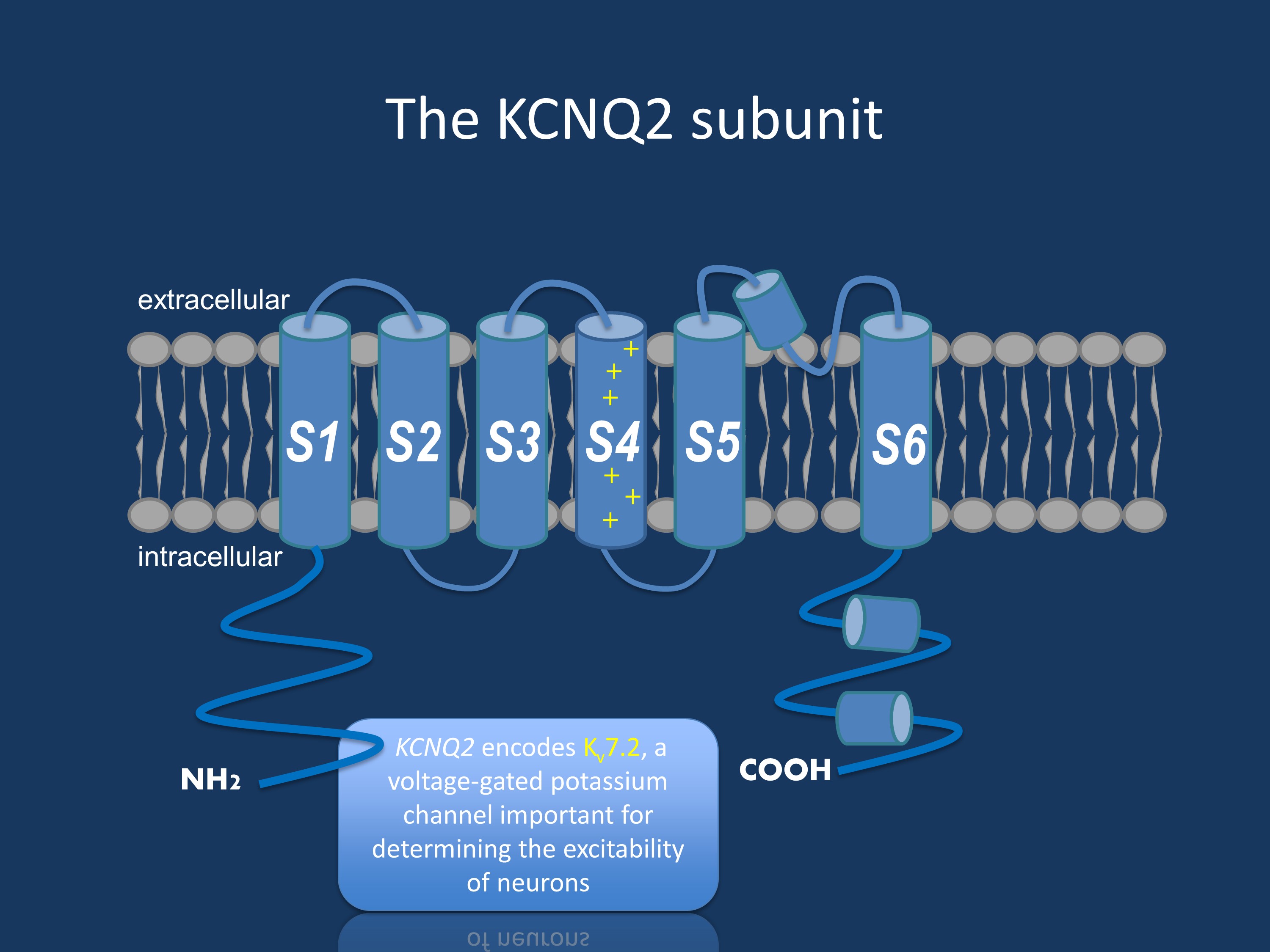
The KCNQ2 subunit. KCNQ2 encodes Kv7.2, a voltage-gated potassium channel important for determining the excitability of neurons
Phenotypes
Overview. There are two main categories of patients that carry mutations in the gene KCNQ2, namely 1) patients with benign (familial) neonatal epilepsy (BFNE), and 2) patients with neonatal epileptic encephalopathy, also called KCNQ2 encephalopathy. Both syndromes make up the two ends of a spectrum, as the developmental problems associated with KCNQ2 mutations can range from absent, over mild to profound intellectual disability. Finally, a small minority of patients with KCNQ2 mutations has a seizure onset during early infancy, under the form of infantile spasms.
Benign familial Neonatal Epilepsy (BFNE). This syndrome was one of the first familial epilepsy syndromes to be described back in 1964, and the genetic cause was subsequently identified in 1998 with the description of mutations in KCNQ2 and later also KCNQ3 in 60-70% of families with BFNE. Since then, many families and a few sporadic patients with benign neonatal seizures and mutations in KCNQ2 have been identified. The syndrome is characterized by neonatal seizures with onset in the first week of life in an otherwise healthy child. Seizures are generally brief, consist of tonic, clonic, apneic or autonomic symptoms, and disappear spontaneously during the first year of life. Children with BFNE show normal development. Around 10% of individuals develop epilepsy later in life though, and a few families with childhood and adolescence onset myokymia, a result of peripheral nerve hyperexcitability, have been reported.
KCNQ2 related neonatal epileptic encephalopathy. More recently, de novo mutations in KCNQ2 have been described in children with a much more severe phenotype. As in BFNE, neonates present with early onset, often tonic, seizures, but they are much more difficult to control, and a delay in development soon becomes apparent. Also, unlike BFNE, the interictal EEG is highly abnormal, showing a burst suppression pattern or multifocal epileptic activity. Some children belonging to the more severe end of the spectrum can present clinical similarities with Ohtahara syndrome. Although seizure frequency decreases in time, and many patients eventually become seizure free, these children have a poor developmental outcome with intellectual disability ranging from mild to profound.
KCNQ2 and infantile spasms. A few patients have been described with de novo KCNQ2 mutations and infantile spasms with onset in the first year of life. It is not known yet whether the mutations that give rise to this later onset phenotype act through a different mutational mechanism, or whether the genetic background of patients plays a role.
Genotype
Overview. Carrying a KCNQ2 variant can have very broad range of consequences, from self-limiting neonatal epilepsy to a severe developmental disorder. Although it is not always possible to predict the phenotype from the genotype, some general genotype-phenotype correlations have become apparent. When viewed in the context of individual clinical histories, these correlations can provide helpful guidance in the clinical interpretation of variants.
BFNE, KCNQ2 encephalopathy. BFNE mutations are most often inherited, and a family history of neonatal seizures is often present. Mutations found in patients with a more severe phenotype mostly occur de novo. In unusual instances, they are inherited from a mosaic parent with a BFNE phenotype. Secondly, all types of mutations are seen in BFNE, ranging from missense mutations, over frameshift and nonsense mutations, to whole gene deletions. In KCNQ2 encephalopathy on the other hand, in nearly all published cases the variant is a missense mutation. As explained below, this is probably related to the functional effect of the respective mutations; in general, KCNQ2 encephalopathy mutations seem to have a greater or different effect on channel function than BFNE mutations. Lastly, especially in KCNQ2 encephalopathy, many recurrent mutations have been identified, the p.A294V mutation being among the most frequent ones. When a variant recurs in unrelated individuals and the clinical features suggest encephalopathy, a diagnosis of KCNQ2 encephalopathy can be made with greater confidence, even in the absence of segregation information. By contrast, interpretation of novel missense variants is less certain, especially if encephalopathy is not severe, other clinical risks or present (i.e, perinatal hypoxia-ischemia), or the phenotype is atypical for KCNQ2 (i.e., infantile spasms). The list of known recurrent mutations in KCNQ2 encephalopathy is currently being collected and will be added to this Epilepsiome page once it has been reviewed and finalized.
Mechanism
Potassium channel. KCNQ2 encodes for a subunit of a voltage-gated potassium channel, also known as Kv7.2, which is mainly expressed in the neurons. Together with KCNQ3, a close homologue, it forms heterotetrameric channels that are responsible for the so-called M-current. This current plays a critical role in determining the excitability of neurons and their response to synaptic input. Mutations leading to BFNE have been studied extensively in vitro, and a 25% loss of the M-current seems sufficient to cause a BFNE phenotype. Only a handful of KCNQ2 encephalopathy mutations have been studied so far, and several of them seem to lead to a greater or almost complete loss of the M-current due to a dominant negative effect of the mutations. Interestingly, a few mutations such as the R201C mutation were shown to lead to a gain of function of the channel. Additional research is needed to study the effects of all different KCNQ2 mutations, and this seems particularly important since such an opposite effect on channel functioning might theoretically have therapeutic consequences. Indeed, the potassium channel opener retigabine has been proposed as a potential targeted therapy for KCNQ2 encephalopathy. But not only do we need more data on the clinical efficacy of this drug, patients with gain of function mutations might also benefit from different treatments than patients with loss of function mutations.
Community
Connection. A KCNQ2 community has emerged, including very active patient organizations (see http://kcnq2.org and http://www.kcnq2cure.org). Members are working on a curated KCNQ2 database, where clinical and genetic information of patients diagnosed with KCNQ2 related disorders is collected anonymously and considered in the context of functional information on the variant when available. The aim of this database is to aid in understanding and clinical interpretation of KCNQ2 variants. Both parents and treating physicians (after consent of parents) can include patients in this registry. The website, including both synopses of known variants and information on how to participate will be online soon (www.rikee.org).