
Mutations, but not germline. Many of the genetic alterations that we aim to investigate within the EuroEPINOMICS projects are so-called germline mutations. In the case of de novo events, these mutations have occurred in the germ cells themselves or in very early development. In the case of autosomal dominant or recessive inheritance, the mutations have been transmitted from parents. In either case, the mutation can be found in every cell of the body. Cancer research is mainly focussed on somatic mutations, which give rise to malignant transformation in already differentiated tissues. In fact, many of the techniques that we currently use in neurogenetics were developed to study somatic genetic aberrations. Array comparative genomic hybridization for example, had initially been established for these purposes before expanding the focus to germline microdeletions and microduplications. While the role of somatic mutations in cancer research is well established, the role somatic rather than germline genetic alterations play in other disorders is mainly speculative. Some initial evidence for somatic point mutations has recently been found in Proteus syndrome, a rare overgrowth syndrome. Activating somatic mutations in AKT1 have recently been identified in this disorder. A recent paper by Lee and colleagues now identifies mutations in several genes in the mTOR pathway in patients with hemimegalencephaly, a severe form of brain malformation. Continue reading
Monthly Archives: June 2012
{kind=link}
Why CNS disorders are more likely to be monogenic
Once again, the flood of rare variants. Deep sequencing studies have revealed an unexpected plethora of rare variants, i.e. genetic variants that can only be found in few or even single individuals. While the genetic architecture of more common genetic variants, so-called Single Nucleotide Polymorphisms (SNPs) is well known through the HapMap project, the role of rare variants identified with recent sequencing studies is difficult to interpret. Basically, for an individual variant it is difficult to establish whether this variant is disease-causing or disease-related based on the frequency in cases. Establishing association at the same level of statistical significance as required for SNPs is difficult given that much larger samples are needed. Furthermore, protein prediction algorithms have their limitations and might not be able to discriminate an accidental from a causal variant, given that every individual might be homozygous or compound homozygous for gene-disrupting variants in at least three genes. We are drowning in a flood of rare variants and cannot distinguish pathological from benign variants very well yet. Continue reading
Old friends
The functional interactions of two genes can be predicted by their conserved proximity in the genomes of distant species. The observation can be used to build large scale networks for bacterial species e.g. in the STRING database but there is little evidence for such conservation in larger eukaryotic species such as animals. Metazoan gene order is scrambled after short periods of evolutionary time and few interactions can be found except for the conserved Hox gene clusters.
Gene-gene pairs in metazoan genomes. Irimia et al. now show the prediction of 600 gene-gene interactions in human and more in other species by analysis of conservation across 17 metazoan genomes and demonstrate the validity by a variety of large scale experiments. In brief, some gene-gene-pairs are more conserved than expected, suggesting a functional relationship. Not all gene pairs are adjacent – longer range interactions are also studied. It’s funny to read such a seemingly simple analysis in 2012 as so many people will have tried similar lines of research after the observations by Abachi and Lieber about bidirectional promoters, i.e. promotors, which affect gene expression in the upstream and downstream direction. The small number of available metazoan genomes might have been a cause for the late discovery. Or am I expecting science to move too fast?

Adachi and Lieber found that bi-directional gene pairs are conserved in higher eukaryotes and suggest the accepted explanation that a single promoter drives the expression of both genes.
Location, location, location. The number of new interactions identified by Irimia et al. is small but the experimental data lined up supposedly point towards high degree of true positives. The identified genes might not be of direct interest to epilepsy genetics as they are primarily found it basic cellular functions. But the observation that conservation is strong on a few gene pairs hopefully allows a glimpse on what shapes the genetic architecture, suggesting other neighbouring genes in humans might have positional effects. A recent publication by Campbell et al. provides an interesting example for epilepsy research and suggests cis-regulatory effects between epilepsy genes at the chromosomal region 9q34 including STXBP1 and SPTAN1. I wonder what role non-coding RNAs play in the cases presented by Irimia et al., which is not touched upon.
How human evolution has shaped epilepsy genes
Man and ape. Comparative genomics is relating the differences between species in the genome to the phenotype. When the first comparisons between human and chimpanzee were published in 2005, neuroscientists were excited that this comparison would show what makes our brain human. A glance at the genes clusters that rapidly evolved during human-specific evolution was sobering. Many of the genes that rapidly evolved are immune-regulatory genes, highlighting our constant struggle with some parasites, bacteria and viruses and our arrangement with the microbiome, the cloud of microbes contribute ten times as many cells to our bodies than we do. Prominent genes such as FOXP2, a gene found to be disrupted in patients with developmental language problems, and most other brain genes barely show up as a group in comparative genomics.
Segmental duplications and human evolution. Recent human evolution did not only leave traces at the single-base pair level that allows for the discovery of individual sequence differences but also larger structural genomic variants including segmental duplications. Recurrent microdeletions such as the 15q13.3, 16p13.11 or 15q11.2 microdeletions occur relatively frequently because the genomic sequence of these deletions is located between segmental duplications (Figure 1). These segmental duplications are so similar that the DNA replication machinery sometimes mistakes one duplication for the other (e.g. the “right flanking duplication” for the “left flanking duplication”). Thereby, the intervening sequence is either deleted or duplicated. Segmental duplications are relatively human-specific, for example a paralogue region for the 15q13.3 microdeletion is not present in rodents. Even in chimpanzees, the segmental duplications that give rise to Prader-Willi-Syndrome, Williams-Syndrome or Spinal Muscular Atrophy are not present, indicating that something very human-specific must have happened here that resulted in these segmental duplications, which give rise to so-called genomic disorders. While many of the epilepsy genes identified through family studies including SCN1A or GABRG2 are conserved in animals, microdeletions are not.

Figure 1. The human genome is a complicated meshwork of duplications and deletions that are lineage-specific, i.e. only occuring in human. One example of this is the genetic architecture of human chromosome 16. When comparing baboon and man, the human chromosome 16 is a complicated puzzle of deletions, duplication, duplications-within-duplications etc. This is the reason why there are at least five different microdeletions syndromes on chromosome 16, resulting from accidental genomic rearrangements between these duplications. This image is modified from a figure on the web page of the Eichler lab.
The SRGAP2 story. Two recent publications on the SRGAP2 gene illustrate the problems with reasoning around the human-specific changes. Saitsu and colleagues describe a female patient with epileptic encephalopathy and a balanced translocation in SRGAP2, a gene highly expressed in the developing forebrain. Balanced translocations are very rare and might provide insight into monogenic epilepsy syndromes. The function of SRGAP2 is still unknown. The second paper by Dennis and collaborators describes the evolution of the SRGAP2 family, hypothesizing that the evolution paralleled neocortical expansion at the transition of Australopithecus to Homo.
How human is our SRGAP2? While the suggestion that disruption of a human-specific gene results in West Syndrome carries some appeal, comes with an almost philosophical flaw. Epileptic encephalopathy, a severe neurodevelopmental disease, may be conceptualized as a disruption of basic cellular or network processes, e.g. large malformations, neurometabolic disorders or fundamental disruptions of synaptic function rather than defects in the subtle differences between man and ape that make us human. In fact, the Toronto Database of Genomic Variants lists several intragenic deletions and duplications in SRGAP2, suggesting that disruptions of this gene may be seen in apparently healthy individuals. Possibly, the genetic architure in SRGAP2 might predispose to deletions, duplications or more complicated rearrangements that even result in balanced translocations. Evolution has merely generated a complex human-specific gene prone to recombination events rather than a genuine epilepsy gene.
Evolutionary genomics and EuroEPINOMICS. So-called hotspot microdeletions including variants at 15q13.3, 16p13.11, 15q11.2 are the immediate result of recent genomic changes that allowed us to be human. Segmental duplications rather than changes on the base-pair level might have been the main driver for human evolution, and the occurence of microdeletions may be conceptualized as a “trade-off” for the fragile human genomic architecture. Microdeletions already help us explain approximately 3% of cases of epilepsy. It can be hypothesized that similar effects on a smaller genomic level are still to be discovered and may help explain additional genetic findings seen in epilepsy patients.
Big data now, scientific revolutions later
Sequence databases are not the only repositories that see exponential growth. The internet helps companies to collect information in unprecedented orders of magnitude, which has spurned the development of new software solutions. “Big data” is the term that stuck with it and blew life into the data analysis. Widespread coverage ensued, including a series of blog posts published by the New York Times. Data produced by sequencing is big: Current hard drives are too slow for raw data acquisition in modern sequencers and we have to ship the discs because we lack the bandwidth to transmit the data via the internet. But we process them only once and in a couple of years from now they can be reproduced with ease.
Large-scale data collection is once again hailed as the next big thing and spiced with calls for a revolution in science. In 2008, Wired even announced the end of theory. Experimental scientists make good use of hypotheses and targeted experiments under the scientific method the last time I checked though. A TEDMED12 presentation by Atul Butte, bioinformatician at Stanford is symptomatic in it’s revolutionary language and caused concern with Florian Markowetz, bioinformatician at the Cancer Center in Cambdridge, UK (and a Facebook friend of mine). Florian complains and explains that the quantitative changes in the data does not lead to a new quality of science and calls for better theories and model development. He’s right, although the issue of data acquisition and source material had deserved more attention (what can you expect from a mathematician).

The part of the data we care about in biology is quite moderate but note that the computing resources of the BGI are in league with the Large Hadron Collider.
We don’t know what to expect from e.g. exome sequencing for a particular disease and the only way to find out is to do the experiment, look at the data, come up with guestimates and confirm your finding in the next round. Current data gathering and analysis projects in the life sciences won’t be classified as big data by the next sweep of scientists anyway. They are mere community technology exploration projects using ad hoc solutions.
Exome sequencing corrects diagnosis in autosomal recessive disorders
The amazing powers of exome sequencing – a disclaimer. We have recently blogged frequently on the power of exome sequencing in monogenic disorders. Dixon-Salazar now describe in “Exome Sequencing Can Improve Diagnosis and Alter Patient Management” the usefulness of exome sequencing in disease identification in autosomal recessive disorders. Their overall yield is a novel gene discovery in 22/118 probands and a different diagnosis than the initial in 10/118 patients. While title and abstract suggest that exome sequencing is a cure-all improving patient diagnosis and altering patient management, it should be pointed out that this manuscript exclusively deals with autosomal recessive disorders. Only two novel genes out of 20 are described, leaving the reader with little chance to investigate their claim. Many of their families were selected from countries with a high consanguinity including Morocco, where state-of-the-art diagnostic facilities are difficult to access for some patients. The only change in patient management resulting from the altered diagnosis was stopping supplementary Vitamin E in a family with a SPG11 mutation previously thought to have ataxia with vitamin E deficiency. What the altered direction of therapy in a family with a newly identified a-mannosidase type 1 entails, is left for the reader to imagine. The corresponding reference refers to a paper on stem cell transplant as a definitive treatment option, which will probably not be a treatment option for this family from Islamabad, Pakistan. The paper rather shows that exome sequencing is of use in autosomal recessive disorders and might yield surprises. Continue reading
One fish, two fish, red fish, blue fish – KCTD13 and neurogenetic studies in zebrafish
Microdeletions in seizure disorders. In a recent paper in Nature, Golzio and colleagues identified KCTD13 as the main driver for the neurodevelopmental phenotype of the 16p11.2 microdeletion. Small losses of chromosomal material as found in microdeletions usually affect several neighbouring genes. Many deletions are due to the particular duplication architecture of the human genome and are canonical, i.e. they always have the same size and include the same genes. The same duplication architecture also makes these variants relatively common, and the full impact of microdeletion-associated genetic morbidity has startled the neurogenetics. The recent five years have led to the identification of several epilepsy-related microdeletions including variants at 15q13.3, 16p13.11 and 15q11.2. There are further microdeletions that are usually found in patients with autism or intellectual disability and to a lesser extent in patients with epilepsy. The 16p11.2 microdeletion, the first microdeletion to be identified through a large-scale association study, is one of these variants.
From deletion to causative genes. For many microdeletions, the statistical evidence for the association with a particular phenotype is often beyond reasonable doubt given that several thousands samples can be included nowadays. The identification of the underlying causative gene, however, is extremely difficult. It is technically challenging and time-consuming to investigate all included genes functionally through conventional model systems. The function of many genes included in microdeletions are not related to ion channels, the best known pathological substrate in epilepsies, and hampers testing effects through established electrophysiological techniques. Finally, microdeletions only lead to hemizygosity, i.e. the second copy of a gene should still be expressed at lower level, requiring model system looking for a quantitative rather than qualitative change. The bottom line is that epilepsy researchers are stuck without suitable model systems, which would allow for a medium-size throughput screening for genes in these deletions. This is where Danio rerio comes into play.
The zebrafish as a model for neurodevelopmental disorders. The zebrafish (Danio rerio) is a good model system for genetic and developmental research. The technologies for genetic manipulation are highly advanced. In addition, embryos are transparent and develop externally. Furthermore, a zebrafish develops quickly and produces a large number of offspring. For her studies on developmental genetics using the zebrafish as a model system, Christiane Nüsslein-Volhard received the Nobel Prize for Medicine in 1995.
Screening of the candidate genes of the 16p11.2 microdeletion. Golzio and coworkers focussed on a peculiar aspect of the 16p11.2 microdeletion as an outcome parameter for their genetic screening – macrocephaly, i.e. an enlarged head circumference. In contrast, patients with the corresponding 16p11.2 microduplication often show microcephaly, i.e. a reduced head circumference. Golzio and colleagues deviced a system to measure head circumference in zebrafish embryos and then overexpressed the 29 genes contained in the 16p11.2 microdeletion in the developing embryo. Strikingly, only KCTD13 resulted in microcephaly. Macrocephaly was seen when KCTD13 was knocked-out with a morpholino. This demonstrated that up- or downregulation of KCTD13 affects head size. The authors went on to show that these differences in head size are driven by differences in neuronal proliferation. KCTD13 is highly expressed in the human forebrain and recent studies have suggested a role for excessive neurons in the frontal lobe in autism.

Figure 1. Study design by Golzio and coworkers to identify KCTD13 as the main gene within the 16p11.2 microdeletion responsible for micro- and macrocephaly. Neuronal proliferation or apoptosis underlies this phenomenon.
Application to epilepsy research. The authors combine a clever screening strategy with a convincing follow-up study, highlighting the potential of zebrafish studies in neurogenetics. However, head circumference is not identical with autism and only represents a surrogate parameter. Therefore, even though the authors emphasize the role of head circumference as an essential part of the 16p11.2 phenotypes, it only represents a minor aspect of it. Nevertheless, the authors demonstrate that Danio rerio is a good model system for medium-throughput screening strategies, and epilepsy models in zebrafish do exist, suggesting that this study design might help decipher the plethora of candidate genes arising from the genetic studies in EuroEPINOMICS.
No use in studying gene-gene and gene-environment effects in complex diseases?
Genome-wide association studies (GWAS) have improved our insight into the genetics of complex diseases but have fallen short of initial expectations, leaving the majority of the heritabililty to be explained. Interactions of genes with the environments and with each other receive a fair share of the blame for the lack of progress despite the widespread efforts. The large number of possible interactions, however, currently still limits progress in this field. A dedicated and growing group of computer scientists and geneticists now study gene-gene effects in the hope of shedding light on complex diseases. Initial results were hopeful, even in the field of epilepsy genetics.
Now, a group of Harvard based biostatisticians presented simulations for breast cancer, type 2 diabetes and rheumatoid arthritis that include gene-gene and gene-environment effects. Their interpretation reads bleak: little predictive power can be gained by including the additional dependencies, which means that all the CPU time consumed currently for their analysis is only warming the planet and the hearts of computer scientists.
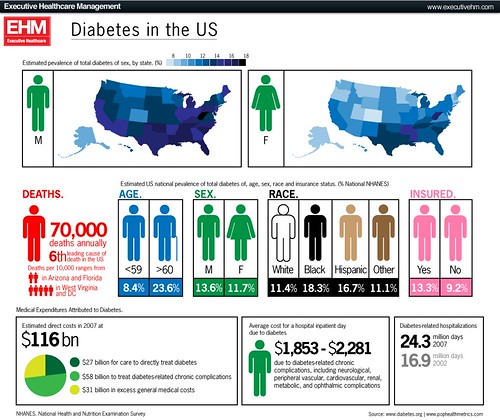
The large number of cases diabetes and many other complex widespread diseases are not explained easily. And the Aschard study suggests that it will remain so for the immediate future despite the progress in sequencing technology.
Negative predictions from experts for their own domain usually receive a negative backlash. The study could probably be attacked on the grounds that the authors selected a large number of parameters, some from probably little more than thin air. But the geneticists on twitter remained silent. Is this acceptance already? Maybe the critics still lie exhausted from attacking Vogelstein’s negative predictions from a couples of months ago.
If the statistical model and parameter choices find widespread acceptance, it would mean that it is virtually impossible to explain many complex diseases from genetics alone to a sufficient degree. As individual studies of the interactions of two SNPs are difficult enough, many cases of complex diseases will remain unexplained. Despite all the efforts, it would be almost as dark as before we had high-throughput sequencing facilities.
FAME – when phenotypes cross over but chromosomes don’t
Crompton and colleagues recently published the clinical and genetic description of a large family with Familial Adult Myoclonic Epilepsy (FAME). This phenotype is particularly interesting since it provides some insight into how neurologists conceptualize twitches and jerks. It is also a good example that large families do not necessarily result in a narrow linkage region, particularly when centromeric regions are involved.
What is myoclonus? Despite usually mentioned in the context of epilepsy, most people are inherently familiar with myoclonus. Most of us “twitch” when we fall asleep and sometimes experience this twitch as part of a dream. These episodes are entirely normal and are called hypnic jerks, but they give people a good idea of what a sudden, brief, shocklike, involuntary movement caused by muscular contraction or inhibition would feel like. Myoclonus in the setting of epilepsy is usually mentioned as part of a Juvenile Myoclonic Epilepsy (JME) or Progressive Myoclonus Epilepsy (PME). Please note that both epilepsies use different endings to describe the twitch (“-us” vs. “–ic”). This is mainly convention. Basically, myoclonus is a brief shock-like twitch, which can affect almost every part of the body and can be due to dysfunctions in various regions in the Central Nervous System.
The neuroanatomy of twitching. A motor command from the cerebral cortex has to pass through several steps prior to execution. For example, the simple command of tapping a finger on the table surface is prepared by the cortex through several loops before being sent down your spine. Accordingly, myoclonus can arise from different parts in the brain. (1) The cortical myoclonus is due to a purely cortical source and can be seen in many forms of symptomatic myoclonus. (2) The cortico-subcortical myoclonus is due to feedback from the cortex to other brain areas. This is the myoclonus we see in patients with JME. Both variants may be seen on EEG since the cortex is involved. (3) The subcortical-supraspinal myoclonus is generated in the brain stem or below and is responsible for phenomena such as hyperekplexia or startle disease. Some forms of hyperekplexia, literally “exaggerated surprise”, are due to mutations in genes involved in glycinergic transmission and can be found in some isolated communities such as the Jumping Frenchmen of Maine. (4) Finally, there is also spinal and peripheral myoclonus.
FAME – epilepsy or movement disorder? Familial Adult Myoclonic Epilepsy (FAME) is an enigmatic familial disorder with the triad of myoclonus, tremor and seizures. Several families have been described and two loci on 8q23.3-8q24.11 and 2p11.1-q212.2 for FAME have been established. The underlying genes are still unknown. Crompton and colleagues no describe a large six-generation family with FAME in Australia/New Zealand. The familial disease usually starts with tremor in early adulthood in the affected family members, even though a wide range of age of onset is observed. Interestingly, only a quarter of all affected family members had seizures, which is in contrast to previous studies. Therefore, FAME may actually be better characterized as a movement disorder with concomitant seizures rather than a familial epilepsy syndrome. The authors also point out the difficulties distinguishing FAME from the much more common essential tremor (ET). In particular, the well-described response to β-blockers seen in patients with ET can also be observed in some family members.

Figure 1. The candidate gene landscape of the chr2 FAME region. All genes were searched for the number of hits in PubMed for the listed search terms in an automated fashion. As usual in large linkage intervals, only few genes are known in the context of neurological disorders, while most genes are unknown.
The genetics of FAME. Crossovers during meiosis usually lead to a progressive narrowing of the linkage interval in familial disorders. However, the lack of crossover events leads to very large linkage intervals even in very extended families. The family described by Crompton et al. links to the pericentromeric region of chromosome 2. Pericentromeric regions usually have a low frequency of crossover events, and this phenomenon has also delayed the identification of other familial epilepsies such as Benign Familial Infantile Seizures with mutations in PRRT2. The linkage region contains almost 100 genes and Figure 1 shows the “candidate gene landscape” in this region. While some genes clearly classify as top candidate genes, the majority of the genes in this region are unknown in the context of epilepsy. Therefore, identification of the FAME gene will be exciting and provide us with novel insight on how genetic alterations may produce combined neurological phenotypes.
Be literate when the exome goes clinical
Exomes on Twitter. Two different trains of thoughts eventually prompted me to write this post. First, a report of a father identifying the mutation responsible for his son’s disease pretty much dominated the exome-related twittersphere. In Hunting down my son’s killer, Matt Might describes his family’s journey that finally led to the identification of the gene coding for N-Glycanase 1 as the cause of his son’s disease, West Syndrome with associated features such as liver problems. The exome sequencing that finally led to the discovery was part of a larger program on identifying the genetic basis of unknown, putatively genetic disorders reported in a paper by Anna Need and colleagues, which is available through open access. This paper is an interesting proof-of-principle study that exome sequencing is ready for prime time. Need and colleagues suggest exome sequencing can find causal mutations in up to 50% of patients. By the way, a gene also that turned up again was SCN2A in a patient with severe intellectual disability, developmental delay, infantile spasms, hypotonia and minor dysmorphisms. This represents a novel SCN2A-related phenotype, expanding the spectrum to severe epileptic encephalopathies.
The exome consult. My second experience last week was my first “exome consult”. A colleague asked me to look at a gene list of a patient to see whether any of the genes identified (there were 300+ genes) might be related to the patient’s epilepsy phenotype. Since I wasn’t sure how to best handle this, I tried to run an automated PubMed search for combination of 20 search terms with a small R script I wrote. Nothing really convincing came up except the realisation that this will be an issue that we will be increasingly faced in the future: working our way through exome dataset after the first “flush” of data analysis did not reveal convincing results. Two terms that came to my mind were bioinformatic literacy as something that we need to improve and Program or be Programmed, a book by Douglas Rushkoff on the “Ten commands of the Digital Age”. In his book, he basically points out that in the future, understanding rather than simply using IT will be crucial.
The cost of interpretation is rising. The Genome Center in Nijmegen suggests on their homepage that by the year 2020, whole-genome sequencing will be a standard tool in medical research. What this webpage does not say is that by 2020, 95% of the effort will not go into the technical aspects of data generation, but into data interpretation. For biotechnology, interpretation will be the largest marketing sector.

By 2020, probably more than 10 million genomes will have been sequenced. Data interpretation rather than data generation will represent the most pressing issue.
So, what about epilepsy? “50% of cases to be identified” sounds good for any grant proposal that I would write, but this might be a clear overestimate. Need and colleagues used a highly selected patient population and even in the variants they identified, causality is sometimes difficult to assess. We are maybe much further away from clinical exome sequencing in the epilepsies than we would like to admit. The only reference point we have for seizure disorders to date is large datasets for patients with autism and intellectual disability. While some genes with overlapping phenotypes can be identified, we would virtually be drowning in exome data without being capable of making sense of this.
10,000 exomes now. I would like to predict that after having identified some low-hanging fruits with monogenic disorders, 10,000 or more “epilepsy exomes” would have to be collected before making significant progress. It is, therefore, crucial not to be tempted by wishful thinking that particular epilepsy subtypes necessarily have to be monogenic, as in the case of epileptic encephalopathies or other severe epilepsies. Much of the genetic architecture of the epilepsies might be more complex than anticipated, requiring larger cohorts and unanticipated perseverance.