
Multi-omics. An emerging avenue of research for investigating the underlying architecture of human disease is the development of multi-omics approaches. Integration and analysis of large-scale data generated from genome sequencing alongside other -omics technologies including transcriptomics, proteomics, and metabolomics, enable a more comprehensive and nuanced insight into biological systems that underlie disease. However, in contrast to genomic data, the generation of multi-omics data remains expensive, time-consuming, and is typically limited in large-scale population studies. In a recent publication, Xu and collaborators developed a model predicting >17,000 multi-omic traits from genomic profiles across 50,000 people. Here is a brief review of their paper, with a focus on the relevance of developing multi-omics resources in 2023.
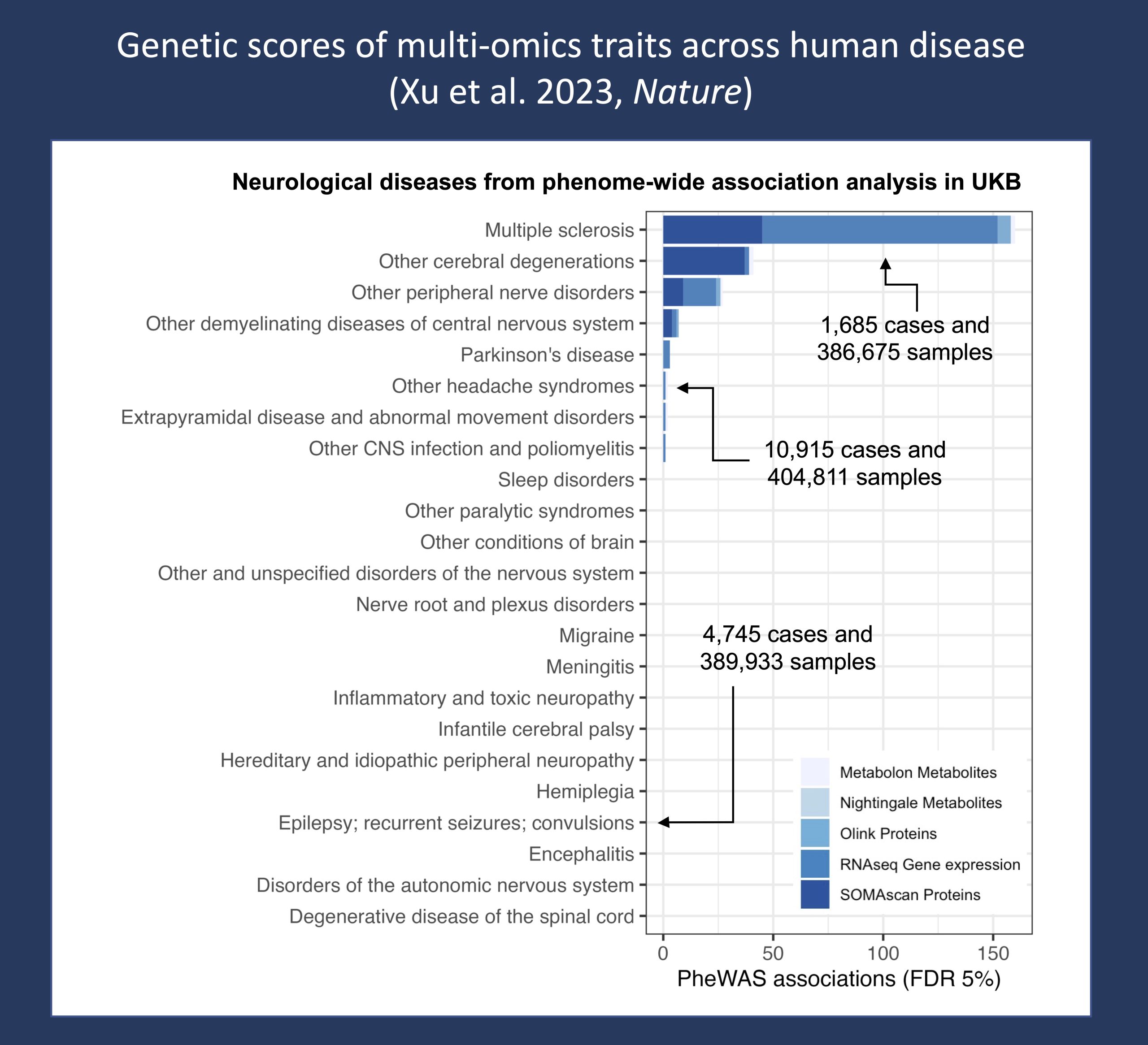
Figure 1. Associations within neurological diseases from a phenome-wide association study (PheWAS) of predicted multi-omic data from the UK Biobank (Xu et al, 2023, Nature). Disease subgroups were defined using PheCodes derived from ICD-9 and ICD-10 diagnosis codes, and only molecular traits with R2 > 0.01 in internal validation were used in the analysis. Multiple sclerosis resulted in the largest number of associations within the broader class of neurological diseases, while the majority of neurological disease subgroups did not result in any significant associations, including epilepsy and seizure disorders. Association data was downloaded from their open-source resource: OmicsPred.
1 – Complexity. Why do we need multi-omics approaches – even in rare disorders where we typically assume the one-gene, one-disease paradigm? While the underpinnings of monogenic diseases are generally understood to be the result of variation in coding regions of single genes, the interplay between genes and their products is typically far more complex and defined by non-linear pathways and networks. For example, the expression of SCN1A is suppressed during the early infantile period due to expression of a “poison exon” that causes nonproductive splicing. Understanding the transcriptional landscape of SCN1A is necessary to explain why the clinical onset of seizures in Dravet Syndrome is typically between 5-6 months of life and the basis of current antisense oligonucleotide therapies targeting nonproductive transcripts. To understand the relevance of multi-omics approaches requires appreciating the complexity of interactions that underlie and define biological systems – moving beyond “one gene, one transcript, one protein.”
2 – Resolution. In the realm of genomic research, multi-omics approaches bridge the gap between genotype and phenotype. Numerous human diseases, including febrile-infection related epilepsy syndrome (FIRES) and multiple sclerosis (MS), have complex and multifactorial etiologies that are likely a combination of genetic, immunologic, and environmental factors. Recognition of downstream molecules and interactions that are more proximal to the biological mechanism can pinpoint pathways and key drivers contributing to disease. For example, following genome-wide association studies, transcriptomics and proteomics are essential in refining signals and implicating causal SNPs with particular disease mechanisms or functional regions. Identification of therapeutic targets hinges on filling in the gap between genotype and phenotype and understanding the “in-between.”
3 – Heterogeneity. In the context of precision medicine, understanding clinical heterogeneity is critical. Why do some individuals with the same genetic variant have different clinical trajectories and outcomes? What drives differences in cancer predisposition and progression rates between types of cancers, between individuals and populations? These knowledge gaps point to opportunities in investigating specific cell and tissue-type omics profiles and provide a rationale for undertaking a systems biology approach to map the molecular signatures of diseases.
Prediction. In their study, Xu and collaborators applied regression analysis to generate multi-omic genetic scores from genomic profiles in a cohort of 48,813 people, predicting the levels of 13,668 RNA transcripts, 2,692 proteins, and 867 metabolites. Through the imputed scores, they were able to characterize the transcriptional landscape of various biological pathways and replicate disease associations in a phenome-wide association study (PheWAS) of predicted multi-omic data from the UK Biobank. What did the study find?
Associations. A total of 18,404 significant associations were identified between genetic scores for multi-omic traits and disease categories, of which, circulatory system, endocrine/metabolic and digestive disorders resulted in the largest number of associations. Notably, integration across multiple omics levels uncovered the role of the JAK-STAT signaling pathway in coronary artery disease and the WNT signaling pathway in hypothyroidism. Less than 2% of associations were linked to neurological disorders (Figure 1), with the most significant association in this disease class between MS and PDE4D, a protein in the phosphodiesterase class – of which selective inhibition has been suggested to promote the regeneration of myelin and thus represents a potential therapeutic target in MS. All associations and predicted genetic scores are available via the open-source resource: OmicsPred. Taken together, Xu and collaborators demonstrate that prediction of multi-omic genetic scores can implicate pathways and biomolecules contributing to disease.
What you need to know. Understanding the complex architecture of human diseases is essential to the identification of molecular targets, development of therapeutics, and application of precision medicine approaches. Multi-omics methodologies are increasingly used in biomedical research to provide comprehensive insight into underlying systems and disease mechanisms. In their recent study, Xu and collaborators predicted genetic scores for multi-omics data from genomic profiles across 50,000 people. Their open-source resource enables researchers to predict molecular traits in their own datasets, accelerating knowledge of complex disease etiologies and providing a foundation for future translation and integration of genetic scores and multi-omics data into the clinical domain.
Julie Xian is a Data Scientist in the Helbig Lab at Children’s Hospital of Philadelphia (CHOP).
