
NDD. Trio-exome sequencing is the gold standard to identify the underlying genetic basis in individuals with neurodevelopmental disorders. De novo variants account for the vast majority of causative genetic findings once a diagnosis is made, but the overall genetic landscape is very heterogeneous, with few genes explaining more than 1% of the genetic morbidity. As the largest study of its kind to date, a recent publication in Nature assessed the spectrum of de novo variants in neurodevelopmental disorders in more than 31,000 individuals. The authors identify more than 250 disease-associated genes, highlight 28 novel genetic etiologies, and highlight signals in their data that hint at more than 1,000 disease-associated genes yet to be discovered. In this blog post, I have summarized the five take-home messages from this large study.
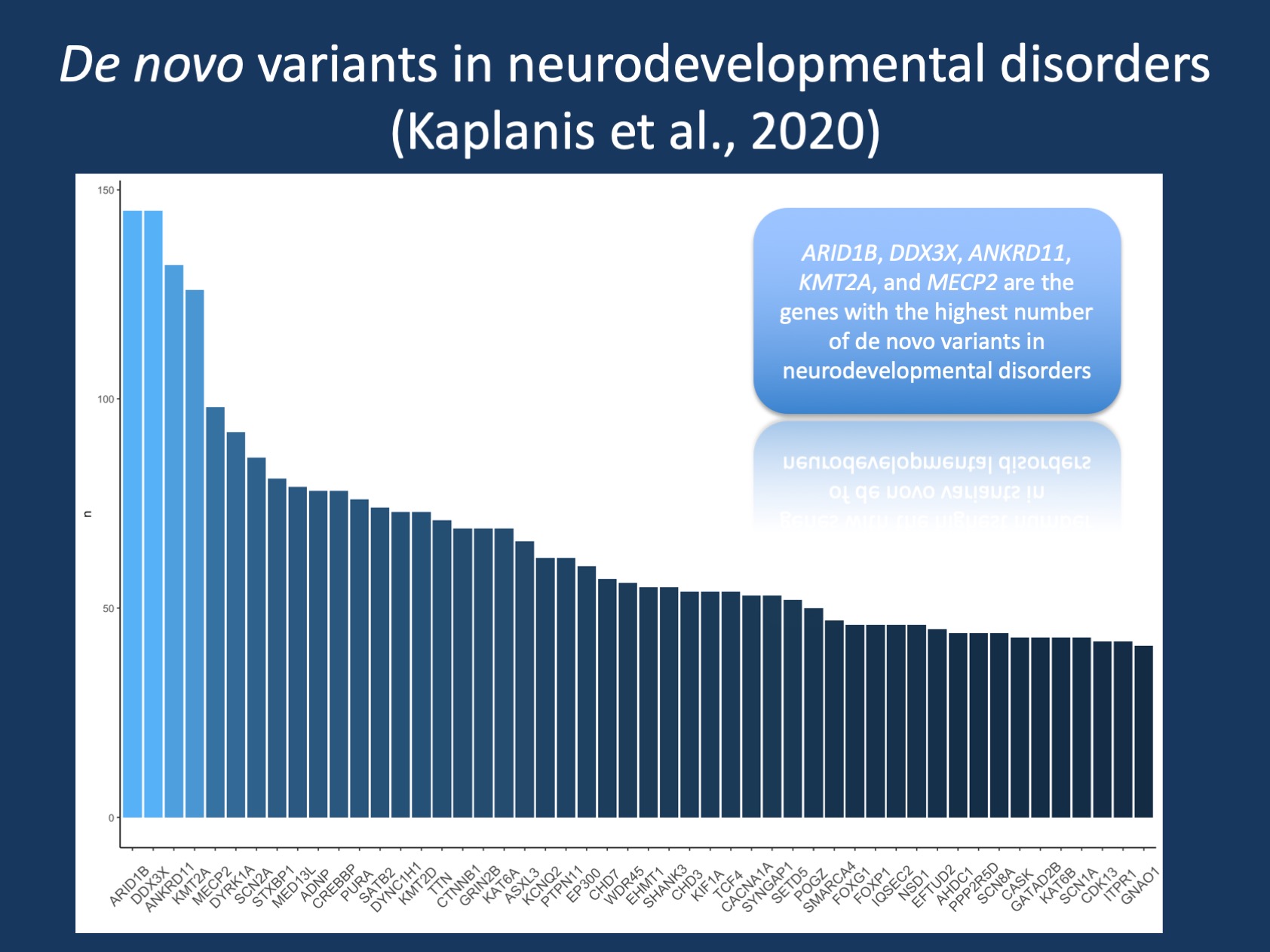
Figure 1. Count of de novo variants for the genes with the most common de novo variants identified by Kaplanis et al. Gene-based count was derived from the Supplementary Data provided by the authors, synonymous de novo variants were excluded, and results were grouped by genes. Common epilepsy-associated genes including SCN2A, STXBP1, KCNQ2, SYNGAP1, SCN8A, SCN1A, and GNAO1 are amongst the 50 most common genes. However, the four most common genes (ARID1B, DDX3X, ANKRD11, and KMT2A) do not represent genetic etiologies that are exclusively associated with the epilepsies and represent broader genetic causes for neurodevelopmental disorders.
De novo variants in 30,000 individuals. To summarize this upfront: the study by Kaplanis and collaborators is enormous, eclipsing prior comparable studies by a factor of three. The authors have analyzed the joint data from three large centers and laboratories, including GeneDx, the DDD study, and Radboud University Medical Center, analyzing the largest group of individuals assessed by trio exome sequencing to date. To further highlight the scope of this study: it is quite possible that most providers reading this blog post will have cared for one or more individuals included in this study. Studies of this magnitude require a somewhat different format than typical research publications, and the main manuscript largely deals with the overall landscape of de novo variants and only mentions a few of the actual genes that the study found. The majority of details on these genes are included in the extensive Supplemental Data. It is quite common for these large-scale publications that the Supplemental Data tells its own story. I have summarized the genes with the highest number of de novo variants as an overview for this blog post (Figure 1). Here are the five take-home messages from this publication.
1 – Identifying NDD genes depends on sample size rather than novel tools
The overall framework to assess whether a specific genetic etiology (e.g. SCN1A, ARID1B) is linked to disease typically occurs by assessing whether de novo variants in a specific gene are more frequent than expected by chance. Going back all the way to the initial Epi4K study, this method has been the gold standard to emphasize that de novo variants in specific genes are not just present, but present at a much higher rate in individuals with disease than expected. The methods used to perform these analyses have changed over the years and the authors present their own novel tool called DeNovoWEST. Using this tool, they provide evidence for a total of 285 NDD genes, including 28 novel genes (link). However, the authors also concede that this large number was largely possible due to the large sample size of 31,058 patient-parent trios and that this novel method only identified 18 more genes than previous methods. Still, improvements in methodology are able to give us a better idea about disease-causing genes. Almost 90% of the genes identified (250/281) had previously been linked to neurodevelopmental disorders either as established (consensus) genes or as candidates considered by at least one center. Overall, 25% of the overall cohort had de novo variants in any of these 281 significantly associated genes.
2 – Novel NDD genes have an unusual frequency of missense variants
Of the 28 novel NDD genes, more than half only had missense variants. Let me quickly highlight why this is important: for most genetic etiologies in NDD, we would assume that haploinsufficiency is the causative disease mechanism. However, for more than half of the novel NDD genes, a different disease mechanism is postulated. For genes including PPP2R5D, PACS2, SRCAP, and CTBP1, the author suggest a dominant-negative mechanism, for PACS1, SMAD4, CSNK2A1, SHOC2, and PTPN11, the authors suggest an activating mechanism. These results highlight that the methods the authors used to identify novel NDD genes are sensitive enough to detect genes with missense variants only and that gain-of-function as a disease mechanism may be relevant to the neurodevelopmental disorders at-large. We had previously suggested that gain-of-function may be relevant for neurodevelopmental disorders with epilepsy, but there may be more to this story. This will be particularly relevant as gain-of-function may require other therapeutic approaches than haploinsufficiency.
3 – Multiple factors account for recurrent de novo variants
Many NDD genes have recurrent de novo variants, but why exactly is this happening? The authors provide compelling evidence that the reasons for recurrent variants are heterogeneous. Some of the recurrence is accounted for by hypermutability, the presence of CpG dinucleotides at specific sites within genes that mutate more frequently. However, this is only one mechanism. Clinical ascertainment is another factor that accounts for de novo variants, as more than half of the recurrent de novo variants were seen in genes that are considered established rather than candidates. Finally, the most cryptic mechanism is positive germline selection, the hypothetical framework that recurrent de novo variants may provide some advantages in the germ cells, while causing disease in the offspring. This mechanism has previously been reported in genes within the RAS-MAPK pathway. Overall, recurrent de novo variants are intriguing, offer a novel insight into disease mechanism, and cannot be exclusively explained by a single factor.
4 – Phenotypic similarity is reduced in novel NDD genes
Our own lab is working on analyses based on phenotypic similarity, and this method also has a brief appearance in the publication by Kaplanis and collaborators. The authors compare the pair-wise phenotypic similarity of novel NDD genes and highlight the fact that individuals with de novo variants in any of the novel genes are less phenotypically similar than individuals with variants in established genes. This finding is quite interesting, highlighting the ongoing importance of genome-first approaches to identify a specific group of NDD genes that have a large phenotypic range. However, I should also emphasize that the methods used by the authors somewhat predispose them to find these genes. To find genetic etiologies that are phenotypically similar, other computational methods would be preferable that are expected to be at least equally powerful in a sample set of this size (see our blog post on Galer et al.).
5 – Hundreds of NDD genes are still to be discovered
Finally, the authors take a look beneath the surface and perform modeling to estimate how many genes might still be out there. For example, they identify an intriguing enrichment of protein-truncating variant in genes with a high pLI, i.e. genes that would not be expected to tolerate protein-truncating variants in unaffected individuals. Through comprehensive modeling approaches, the authors estimate that there are probably approximately 1,000 NDD genes still out there, but that at least some of them may be difficult to find due to incomplete penetrance and pre- or perinatal death. This suggests that identification of genetic causes of NDD will be an ongoing task. However, the genetic etiologies the authors are referring to are not entirely novel findings – these are the de novo variants that we already know, but are unable to fully understand.
This is what you need to know
The study by Kaplanis and collaborators is a milestone – the largest trio exome sequencing study in neurodevelopmental disorders to date. In more than 30,000 individuals, the authors find statistical evidence for more than 280 disease-associated genes in neurodevelopmental disorders, including 28 novel genetic causes. This large-scale study sheds light on the overall genetic architecture of neurodevelopmental disorders and suggests that there are more than 1,000 genes still to be discovered.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
