
SCN2A. This is the Epilepsiome page for SCN2A, one of the most common genetic causes for neurodevelopmental disorders, including epileptic encephalopathies. The phenotypic spectrum ranges from self-limited neonatal/infantile seizures (formerly known as benign neonatal/infantile seizures), to autism/intellectual disability/schizophrenia, infantile spasms, severe early-onset epileptic encephalopathies, and episodic ataxia.
Here are the most recent blog posts that mention SCN2A
- SCN2A – a neurodevelopmental disorder digitized through 10,860 phenotypic annotations
- Five things I learned at the FamilieSCN2A Annual Family and Professional Conference
- Understanding the SCN2A mystery- therapeutic responses in a heterogeneous disease
- Five things I learned about SCN2A in Chicago
- SCN2A – a 2016 Update
In a nutshell. SCN2A is one of the most common causes of neurodevelopmental disease. Phenotypes include self-limited neonatal/infantile seizures, autism/intellectual disability/schizophrenia, infantile spasms progressing to epileptic encephalopathy, severe early-onset epileptic encephalopathy, and episodic ataxia. Movement disorders seem to be common in patients with epileptic encephalopathy. SCN2A encodes an alpha subunit in a voltage-gated sodium channel and is pivotal for neuronal signaling. With the continued efforts in functional characterization in vivo and in vitro, the underlying mechanism for different phenotypic groups is mostly accounted for.
Phenotypes | Genetics | Mechanism | The Clinical Perspective | Community
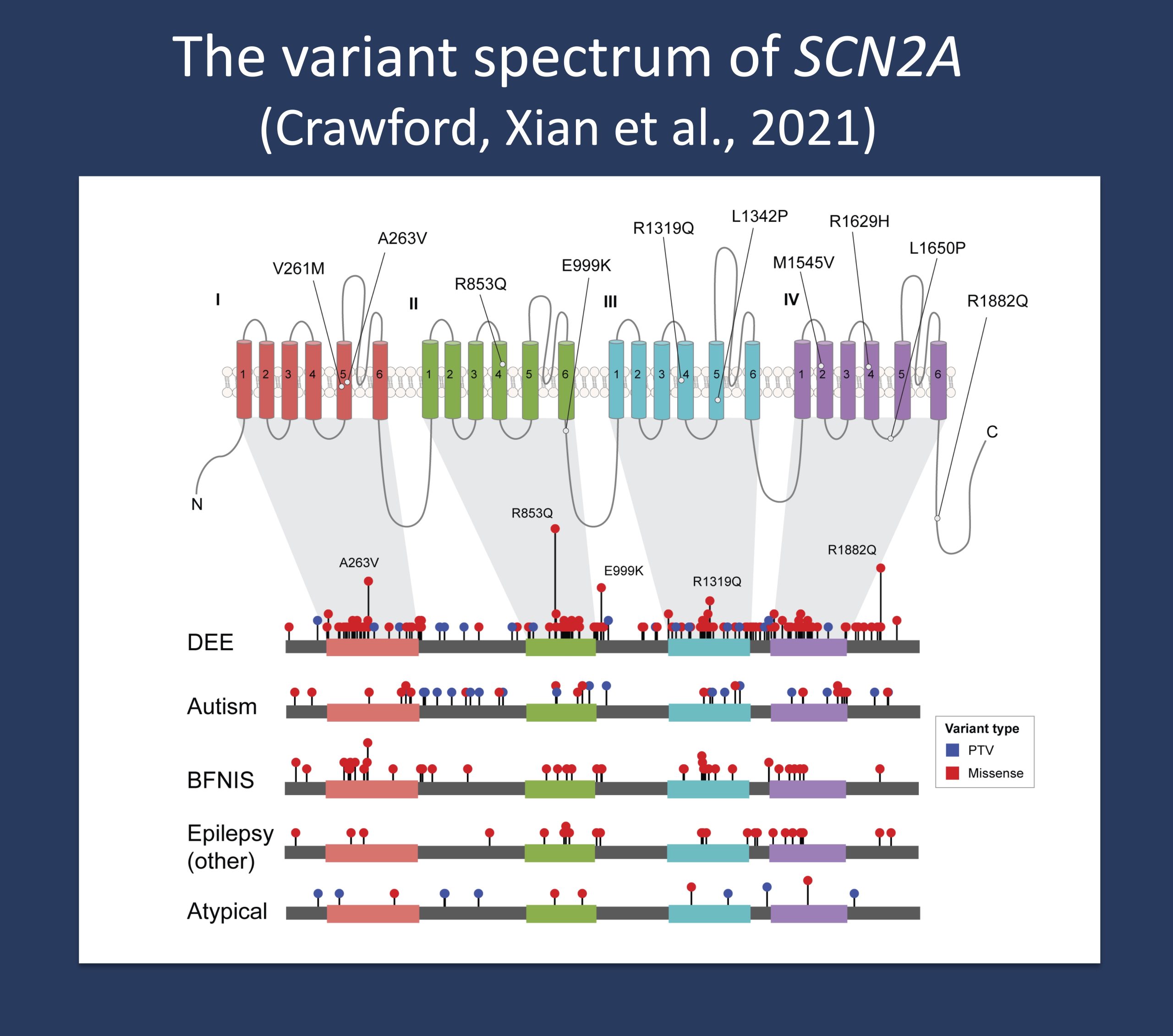
Review of known variants and their associated phenotypes in SCN2A from our 2021 paper by Crawford et al.
Phenotypes
General overview. There is no comprehensive overview of SCN2A phenotypes available to date, and the full spectrum of neurodevelopmental disorders due to variants in this gene is not fully understood. Based on current understanding of the literature, we can breakdown the SCN2A phenotypes into four different groups including (1) self-limited infantile or neonatal seizures, (2) neurodevelopmental/neuropsychiatric disorders, including schizophrenia, autism and intellectual disability, (3) Infantile Spasms, (4) early onset epileptic encephalopathies, including Ohtahara syndrome, Dravet syndrome, West syndrome, epilepsy of infancy with migrating focal seizures and severe neonatal epilepsies, and (5) episodic ataxia. All phenotypes within the SCN2A spectrum can be conceptualized along three dimensions including (a) cognitive outcome, (b) seizures, and (c) movement disorders. Here are the main phenotypes of this spectrum.
Self-limited neonatal/infantile seizures The familial syndrome of benign familial neonatal/infantile seizures was the first SCN2A phenotype to be described, now called self-limited neonatal/infantile seizures. Although this phenotype was the first to be described, it is likely the least common presentation of an SCN2A mutation. Mutations were identified in large families where affected individuals had neonatal and/or infantile seizures and a typical neurodevelopmental outcome. This familial form of SCN2A has been reported in independent studies after the initial discovery, but it became obvious quickly that this familial syndrome is much less common than familial neonatal seizures due to mutation in KCNQ2 or infantile seizures due to PRRT2. Also, this phenotype can be seen in families with SCN8A mutations.
Autism, intellectual disability, schizophrenia. After the initial discovery of SCN2A as a gene associated with BFNIS, it became silent around SCN2A. Interestingly, this gene re-emerged as one of the most obvious candidate genes in many neurodevelopmental disorders in large exome studies. Several studies found de novo variants in SCN2A in patients with neurodevelopmental/neuropsychiatric phenotypes including intellectual disability, autism, and schizophrenia. SCN2A variants associated with these conditions are usually truncating, suggesting that haploinsufficiency plays a major role. Although individuals in this phenotypic group do not typically have neonatal or infantile onset seizures as seen in other groups, later onset seizures can be observed.
Infantile Spasms. We have broken down the epileptic encephalopathy phenotypes caused by variants in SCN2A in two broad categories – epileptic encephalopathies presenting with Infantile Spasms (usually at the age of 3-6 months) or severe epilepsies starting earlier. Even though there is probably a gradient between both phenotypes, we felt that breaking down the SCN2A epileptic encephalopathies into two major categories may make sense from a clinical perspective, as we usually think about different causes for seizures in children presenting with epilepsy as neonates compared to the epilepsy in children developing Infantile Spasms after an initial normal development. The most recurrent SCN2A variant to date, p.R853Q, typically presents with infantile spasms first. Some patients with SCN2A-related Infantile Spasms are also reported to have movement disorders, including later onset episodic ataxia and myoclonus.
Severe early-onset epileptic encephalopathies. There is an entire spectrum of severe epilepsies due to pathogenic variants in SCN2A that start neonatally or within the first three months of life. For some epilepsies, it is possible to classify them into known epilepsy syndromes, such as West syndrome, Ohtahara syndrome, epilepsy of infancy with migrating focal seizures, or Dravet syndrome. For other epilepsies, a classification is more difficult, and these conditions are referred to as unclassified epileptic encephalopathies. Again, movement disorders seem to be common in this group of severe epilepsies. These variants typically have overall “gain-of-function,” with some having mixed functional effects underlying this.
Episodic ataxia. There are individuals in the literature of individuals with episodic ataxia starting in infancy to teenage age, often with early-onset seizures. Cognitive outcome in these individuals tended to be typical.
Other phenotypes. SCN2A variants have also been reported in a small number of patients with acute encephalopathy with biphasic seizures and late reduced diffusion (AESD), but the pathogenicity of at least one of these variants has been questioned. At least one of these reported patients had an unaffected parent who had the same variant, making it unclear whether or not the SCN2A variant was truly disease-causing.
Genetics
Mutational spectrum. There are hundreds of different potential disease-causing variants have been reported in SCN2A to date in the medical literature. Variants have been reported throughout the SCN2A gene. The vast majority of these variants are heterozygous missense variants. Many of these variants have only been reported in a single patient or family, but there are over 60 variants that have been reported in more than one individual. Protein-truncating variants (nonsense, splice site, frameshift,) and some missense variants have also been reported in patients with epilepsy, intellectual disability, and/or autism. Missense variants, some of which have been functionally studied, are the typical variant type for the epileptic encephalopathy phenotypes.
Genotype-Phenotype Correlation. In the most basic framework, variants associated with haploinsufficiency such as frameshift or nonsense variants are typically associated with autism/intellectual disability with the possibility of later onset seizures and “gain-of-function” variants are associated with neonatal-infantile onset epilepsies. The likelihood of developing later-onset seizures in individuals with presumed loss-of-function variants does not seem to be related to the particular variant. Some of the variants seen in epileptic encephalopathies are recurrent, such as p.R853Q or p.R1882Q. Some missense variants can be associated with variable phenotypes. For example, the p.A263V recurrent variant has been reported in cases of epileptic encephalopathy, self-limited neonatal-infantile seizures, episodic ataxia, and autism spectrum disorder. De novo missense and truncating variants are usually associated with a more severe phenotype, while inherited missense variants are usually associated with milder phenotypes, such as self-limited seizures.
Transcripts. Alternative splicing of the SCN2A gene results in two different transcripts, a neonatal (6N) and an adult (6A) transcript, differing by a single amino acid substitution at position 209. There is an asparagine (uncharged) in the neonatal isoform at this position, and this is an aspartate (negatively charged) in the adult isoform. This position is in segment S3-S4 of domain 1. The different charge of the 6N isoform makes the channel less permeable to sodium and therefore makes the cell less excitable. The neonatal transcript is expressed directly after birth and in early postnatal development and shifts during development to the adult transcript. Over development, SCN8A becomes more predominantly expressed over the adult isoform of SCN2A. There have been several cases of disease-causing variants in SCN2A that are present only in the neonatal transcript, which is not necessarily the reference transcript used by diagnostic laboratories. Therefore, if there is a high clinical suspicion of an SCN2A-related condition, sequencing of the neonatal transcript should be considered.
General Considerations for Variant interpretation. When reviewing a genetic variant to determine its significance for a given patient, it is important to weigh multiple pieces of evidence:
Considerations for gene level interpretation. First, it is important to establish the strength of the evidence showing that the gene is associated with epilepsy or other neurodevelopmental phenotypes. Some genes may only have one variant reported in a single individual with epilepsy, while other genes may have multiple variants reported in many large families with an autosomal dominant pattern of epilepsy. For SCN2A, there is very strong evidence for its role in human epilepsy.
Considerations for variant level interpretation. When reviewing the significance of a variant, it is important to consider the impact on the gene and the presence of the variant in previously described patient and control populations. Many clinical genetic testing laboratories classify genetic variants into different categories, ranging from benign to pathogenic. Variants that are common in control populations and would not be predicted to have a major impact on the gene/protein are generally classified as benign. Variants are more likely to be classified as pathogenic if the variants are rare or not present in the control population, reported in multiple individuals or families with disease, and likely to have a higher impact on the gene/protein based on the type of mutation or functional studies. Variants with uncertain or limited available evidence may be classified as variants of uncertain significance (VUS), indicating that further information is required in order for the variant to be further defined. In some cases, testing additional family members can be helpful, as it allows the lab to determine whether or not the variant was inherited (versus de novo) and how the variant segregates with disease in the family. Sometimes further classification of a VUS requires waiting for the identification of additional patients or families with similar or nearby variants. Functional studies or functional prediction models can also assist in variant interpretation or consideration of potential phenotypic outcomes. The considerations for SCN2A variant interpretation are complex given the heterogeneity of phenotypes.
Inheritance, Penetrance & Prevalence. SCN2A variants can be de novo or inherited. With the exception of self-limited neonatal/infantile seizures which are inherited in an autosomal dominant pattern, most SCN2A variants appear to be sporadic and de novo. The penetrance of SCN2A-associated phenotypes appears to be high. Considerable variable expressivity, particularly in seizure type and severity, can occur.
Mechanism & Function
Haploinsuffiency. SCN2A codes for the voltage-gated sodium channel, neuronal type II, alpha subunit. The SCN2A protein is located at the axon initial segment, the key structure in neurons where the decision is made whether the addition of all excitatory and inhibitory inputs will result in the firing of an action potential. Therefore, the function of SCN2A is pivotal to neurons. Truncating variants obviously results in haploinsufficiency for SCN2A as seen in some patients with autism and intellectual disability with or without later onset epilepsy.
“Gain of function.” The functional impact of missense variants, other than variants leading to obvious lack of peak current, can be complex. The simplistic mechanism for variants leading to early-onset phenotypes is “gain of function,” but there are many different functional features underlying this which in many cases have in fact mixed effects on multiple biophysical parameters. This is an important consideration in the development of targeted therapies.
Cell/Animal models.
Mice. An SCN2A mouse model (Q54) presents with seizures and episodic behavioral arrest with stereotypical behaviors, such as tail wagging, head bobbing and chewing. Mice eventually developed frequent spontaneous generalized tonic-clonic seizures that resulted in premature death (Kearney et al, 2001). The SCN2A haploinsufficiency mouse model causes an impaired memory and reduced reactivity to stressful stimuli. However, as the mice reach adult age, the primary notable feature becomes communicative deficits. Other haploinsufficiency models have been used to model sleep disturbances or suggest roles for SCN2A in learning and memory.
The Clinical Perspective
Recurrence Risk & Testing of Family Members. SCN2A variants can be de novo or inherited in an autosomal dominant pattern. If a variant in SCN2A is detected in an unaffected parent, this variant is inherited and may be an innocent bystander, meaning the patient may have a different disease-causing variant in another gene. Some SCN2A variants are recurrent and may therefore be considered pathogenic without parental confirmation. Each child of an individual with a disease-causing variant has a 50% (1 in 2) of inheriting the disease-causing variant and an equal chance of inheriting the functional (wild-type) copy of the gene. Germline mosaicism of an SCN2A variant has been reported, making the recurrence risk for families with a child with a de novo variant with an SCN2A variant higher than the general population risk.
Therapy. There is no gene or gene-modifying therapy recommended for treatment of SCN2A-associated disorders to date. Given the vast genetic and phenotypic heterogeneity of SCN2A phenotypes and the variability of seizure phenotypes even in patients with identical variants, a variety of AED effects have been reported that may not be necessarily related to the underlying disease-causing variant. That being said, sodium channel blockers are a first-line consideration for individuals with presumed or confirmed “gain-of-function.” However, given the expanding number of patients with identified SCN2A variants, identifying gene- or variant-specific drug responses is an active field of research in the epilepsy genetics community.
Community
Family advocacy organizations. The FamilieSCN2A Foundation a non-profit organization for SCN2A families and researchers. They have various resources and support for families and host regular conferences.
Research studies. The scientific community is currently actively studying SCN2A and its role in human disease. The SCN2A Epilepsiome team is happy to facilitate if you have questions or a specific interest in this gene.
Resources & References
Websites
For Families:
SCN2A Genetics Home Reference https://ghr.nlm.nih.gov/gene/SCN2A
References
Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities.
Baasch AL, Hüning I, Gilissen C, Klepper J, Veltman JA, Gillessen-Kaesbach G, Hoischen A, Lohmann K.
Epilepsia. 2014 Apr;55(4):e25-9.
PMID: 24579881
Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy.
Berkovic SF, Heron SE, Giordano L, Marini C, Guerrini R, Kaplan RE, Gambardella A, Steinlein OK, Grinton BE, Dean JT, Bordo L, Hodgson BL, Yamamoto T, Mulley JC, Zara F, Scheffer IE.
Ann Neurol. 2004 Apr;55(4):550-7.
PMID:15048894
Mutation screening of SCN2A in schizophrenia and identification of a novel loss-of-function mutation.
Carroll LS, Woolf R, Ibrahim Y, Williams HJ, Dwyer S, Walters J, Kirov G, O’Donovan MC, Owen MJ.
Psychiatr Genet. 2016 Apr;26(2):60-5.
PMID: 26555645
Novel de novo SCN2A mutation in a child with migrating focal seizures of infancy.
Dhamija R, Wirrell E, Falcao G, Kirmani S, Wong-Kisiel LC.
Pediatr Neurol. 2013 Dec;49(6):486-8.
PMID: 23988467
Autism in several members of a family with generalized epilepsy with febrile seizures plus.
Dixon-Salazar TJ, Keeler LC, Trauner DA, Gleeson JG.
J Child Neurol. 2004 Aug;19(8):597-603.
PMID: 15605469
De novo nonsense mutations in the sodium channel gene, SCN2A, in sporadic intractable epilepsy.
Franciosi S.
Clin Genet. 2010 Jun;77(6):538-40.
PMID: 20236112
A case of recurrent encephalopathy with SCN2A missense mutation.
Fukasawa T, Kubota T, Negoro T, Saitoh M, Mizuguchi M, Ihara Y, Ishii A, Hirose S.
Brain Dev. 2015 Jun;37(6):631-4.
PMID: 25457084
Infantile epileptic encephalopathy, transient choreoathetotic movements, and hypersomnia due to a De Novo missense mutation in the SCN2A gene.
Hackenberg A, Baumer A, Sticht H, Schmitt B, Kroell-Seger J, Wille D, Joset P, Papuc S, Rauch A, Plecko B.
Neuropediatrics. 2014 Aug;45(4):261-4.
PMID: 24710820
The voltage-gated sodium channel gene SCN2A and idiopathic generalized epilepsy.
Haug K, Hallmann K, Rebstock J, Dullinger J, Muth S, Haverkamp F, Pfeiffer H, Rau B, Elger CE, Propping P, Heils A.
Epilepsy Res. 2001 Dec;47(3):243-6.
PMID: 11738931
Hlf is a genetic modifier of epilepsy caused by voltage-gated sodium channel mutations.
Hawkins NA, Kearney JA.
Epilepsy Res. 2016 Jan;119:20-3.
PMID: 26656780
SCN2A mutations and benign familial neonatal-infantile seizures: the phenotypic spectrum.
Herlenius E, Heron SE, Grinton BE, Keay D, Scheffer IE, Mulley JC, Berkovic SF.
Epilepsia. 2007 Jun;48(6):1138-42.
PMID: 17386050
Sodium-channel defects in benign familial neonatal-infantile seizures.
Heron SE, Crossland KM, Andermann E, Phillips HA, Hall AJ, Bleasel A, Shevell M, Mercho S, Seni MH, Guiot MC, Mulley JC, Berkovic SF, Scheffer IE.
Lancet. 2002 Sep 14;360(9336):851-2. Erratum in: Lancet 2002 Nov 9;360(9344):1520.
PMID: 12243921
SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures.
Howell KB, McMahon JM, Carvill GL, Tambunan D, Mackay MT, Rodriguez-Casero V, Webster R, Clark D, Freeman JL, Calvert S, Olson HE, Mandelstam S, Poduri A, Mefford HC, Harvey AS, Scheffer IE.
Neurology. 2015 Sep 15;85(11):958-66.
PMID: 26291284
Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing.
Jiang YH, Yuen RK, Jin X, Wang M, Chen N, Wu X, Ju J, Mei J, Shi Y, He M, Wang G, Liang J, Wang Z, Cao D, Carter MT, Chrysler C, Drmic IE, Howe JL, Lau L, Marshall CR, Merico D, Nalpathamkalam T, Thiruvahindrapuram B, Thompson A, Uddin M, Walker S, Luo J, Anagnostou E, Zwaigenbaum L, Ring RH, Wang J, Lajonchere C, Wang J, Shih A, Szatmari P, Yang H, Dawson G, Li Y, Scherer SW.
Am J Hum Genet. 2013 Aug 8;93(2):249-63.
PMID: 23849776
A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline.Kamiya K, Kaneda M, Sugawara T, Mazaki E, Okamura N, Montal M, Makita N, Tanaka M, Fukushima K, Fujiwara T, Inoue Y, Yamakawa K.
J Neurosci. 2004 Mar 17;24(11):2690-8.
PMID: 15028761
A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH.
Neuroscience. 2001;102(2):307-17.
PMID: 11166117
Scn2a sodium channel mutation results in hyperexcitability in the hippocampus in vitro.
Kile KB, Tian N, Durand DM.
Epilepsia. 2008 Mar;49(3):488-99.
PMID: 18031550
Acute encephalopathy with a novel point mutation in the SCN2A gene.
Kobayashi K, Ohzono H, Shinohara M, Saitoh M, Ohmori I, Ohtsuka Y, Mizuguchi M.
Epilepsy Res. 2012 Nov;102(1-2):109-12.
PMID: 22591750
SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain.
Liao Y, Anttonen AK, Liukkonen E, Gaily E, Maljevic S, Schubert S, Bellan-Koch A, Petrou S, Ahonen VE, Lerche H, Lehesjoki AE.
Neurology. 2010 Oct 19;75(16):1454-8.
PMID: 20956790
Molecular correlates of age-dependent seizures in an inherited neonatal-infantile epilepsy.Liao Y, Deprez L, Maljevic S, Pitsch J, Claes L, Hristova D, Jordanova A, Ala-Mello S, Bellan-Koch A, Blazevic D, Schubert S, Thomas EA, Petrou S, Becker AJ, De Jonghe P, Lerche H.
Brain. 2010 May;133(Pt 5):1403-14.
PMID: 20371507
Confirming an expanded spectrum of SCN2A mutations: a case series.
Matalon D, Goldberg E, Medne L, Marsh ED.
Epileptic Disord. 2014 Mar;16(1):13-8.
PMID: 24659627
Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome.
Nakamura K, Kato M, Osaka H, Yamashita S, Nakagawa E, Haginoya K, Tohyama J, Okuda M, Wada T, Shimakawa S, Imai K, Takeshita S, Ishiwata H, Lev D, Lerman-Sagie T, Cervantes-Barragán DE, Villarroel CE, Ohfu M, Writzl K, Gnidovec Strazisar B, Hirabayashi S, Chitayat D, Myles Reid D, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Hayasaka K, Matsumoto N, Saitsu H.
Neurology. 2013 Sep 10;81(11):992-8.
PMID: 23935176
De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies.
Ogiwara I, Ito K, Sawaishi Y, Osaka H, Mazaki E, Inoue I, Montal M, Hashikawa T, Shike T, Fujiwara T, Inoue Y, Kaneda M, Yamakawa K.
Neurology. 2009 Sep 29;73(13):1046-53.
PMID: 19786696
Missense mutations in sodium channel SCN1A and SCN2A predispose children to encephalopathy with severe febrile seizures.
Saitoh M, Ishii A, Ihara Y, Hoshino A, Terashima H, Kubota M, Kikuchi K, Yamanaka G, Amemiya K, Hirose S, Mizuguchi M.
Epilepsy Res. 2015 Nov;117:1-6.
PMID: 26311622
Severe epilepsy, retardation, and dysmorphic features with a 2q deletion including SCN1A and SCN2A.
Pereira S, Vieira JP, Barroca F, Roll P, Carvalhas R, Cau P, Sequeira S, Genton P, Szepetowski P.
Neurology. 2004 Jul 13;63(1):191-2.
PMID: 15249644
Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia.
Schwarz N, Hahn A, Bast T, Müller S, Löffler H, Maljevic S, Gaily E, Prehl I, Biskup S, Joensuu T, Lehesjoki AE, Neubauer BA, Lerche H, Hedrich UB.
J Neurol. 2016 Feb;263(2):334-43.
PMID: 26645390
Clinical spectrum of SCN2A mutations.
Shi X, Yasumoto S, Kurahashi H, Nakagawa E, Fukasawa T, Uchiya S, Hirose S.
Brain Dev. 2012 Aug;34(7):541-5.
PMID: 22029951
Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome.Shi X, Yasumoto S, Nakagawa E, Fukasawa T, Uchiya S, Hirose S.
Brain Dev. 2009 Nov;31(10):758-62..
PMID: 19783390
A novel SCN2A mutation in family with benign familial infantile seizures.
Striano P, Bordo L, Lispi ML, Specchio N, Minetti C, Vigevano F, Zara F.
Epilepsia. 2006 Jan;47(1):218-20.
PMID: 16417554
A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction.
Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K.
Proc Natl Acad Sci U S A. 2001 May 22;98(11):6384-9. Erratum in: Proc Natl Acad Sci U S A 2001 Aug 28;98(18):10515.
PMID: 11371648
Whole genome sequencing identifies SCN2A mutation in monozygotic twins with Ohtahara syndrome and unique neuropathologic findings.
Touma M, Joshi M, Connolly MC, Grant PE, Hansen AR, Khwaja O, Berry GT, Kinney HC, Poduri A, Agrawal PB.
Epilepsia. 2013 May;54(5):e81-5.
PMID: 23550958
Sodium channels SCN1A, SCN2A and SCN3A in familial autism.
Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, Reichert J, Buxbaum JD, Meisler MH.
Mol Psychiatry. 2003 Feb;8(2):186-94.
PMID: 12610651
Paternal germline mosaicism of a SCN2A mutation results in Ohtahara syndrome in half siblings.
Zerem A, Lev D, Blumkin L, Goldberg-Stern H, Michaeli-Yossef Y, Halevy A, Kivity S, Nakamura K, Matsumoto N, Leshinsky-Silver E, Saitsu H, Lerman-Sagie T.
Eur J Paediatr Neurol. 2014 Sep;18(5):567-71.
PMID: 24814476
The SCN2A Epilepsiome Team
Ingo Helbig
Rikke Moller
Katrine Johannesen