
TBC1D24. This is the Epilepsiome page on TBC1D24. Pathogenic variants in this gene can cause various epileptic disorders (with or without intellectual disability or hearing loss) or non-syndromic hearing loss. With over 30 families identified, it might be one of the more common recessive causes of epilepsy.
Here are the most recent blog posts on TBC1D24
- TBC1D24 – this is what you need to know in 2015
- Publications of the week – Dravet Syndrome, TBC1D24, and CSTB
- Publications of the week – ATK3, TBC1D24, and BRAT1
- TBC1D24, DOORS Syndrome, and the unexpected heterogeneity of recessive epilepsies
- The return of TBC1D24
In a nutshell. Pathogenic variants in TBC1D24 were initially found in two families with different types of autosomal recessive epilepsy in 2010. These reports were followed by the identification of disease-causing variants in another family with recessive epilepsy in 2013, and then by the identification of 9 families with TBC1D24 variants and DOORS syndrome (Deafness, Onycho-Osteodystrophy, mental Retardation and Seizures). Surprisingly, TBC1D24 variants were then also identified in families with autosomal dominant or recessive non-syndromic hearing loss without epilepsy. Thus, pathogenic TBC1D24 variants are associated with a wide phenotypic spectrum.
| Phenotypes | Genetics | Mechanism & Function |
| The Clinical Perspective | Community | Resources & References |
Phenotypes
Pathogenic variants in TBC1D24 can cause a wide range of phenotypes, including many that feature seizures with or without myoclonus:
Familial infantile myoclonic epilepsy (FIME) is characterized by early-onset myoclonic seizures, focal epilepsy, dysarthria, and intellectual disability or developmental delay. Intellect may be normal.
Progressive myoclonus epilepsy (PME) is characterized by action myoclonus, tonic-clonic seizures, progressive neurologic decline, and ataxia. In patients with TBC1D24-related epilepsy, this can present in infancy as episodic multifocal myoclonus with onset of other seizure types not appearing until over five years later.
Early-infantile epileptic encephalopathy 16 (EIEE16) is characterized by epilepsy and epileptiform EEG abnormalities, which could contribute to progressive neurologic decline.
DOORS. The acronym DOORS refers to the combination of deafness, onychodystrophy, osteodystrophy, intellectual disability (mental retardation), and seizures. DOORS syndrome is a rare, autosomal recessive syndrome that has epilepsy as an inconsistent feature. Accordingly, the “S” in DOORS is sometimes omitted (DOOR). Onychodystrophy refers to malformation of the nails, which can be malformed or even absent. Osteodystrophy refers to malformation of the skeletal system. In patients with DOORS Syndrome, usually the hands and feet are affected. One third of patients have a triphalangeal thumb, a thumb with three instead of the usual two joints. The epilepsy in DOORS syndrome usually starts in the first year of life and can be refractory to treatment. Using exome sequencing in 26 families with DOORS Syndrome, Campeau and collaborators identified TBC1D24 variants in 9 families. These findings suggest that the role of TBC1D24 extends beyond the central nervous system and that it is also involved in the formation of bone and skin structures.
Other than the unique constellation of features associated with DOORS syndrome, the other epilepsy phenotypes associated with TBC1D24 variants are variable and not distinctive enough to indicate when TBC1D24 testing is appropriate.
Other features and phenotypes include:
Brain abnormalities. Brain imaging may show cerebellar atrophy with an increased signal of the cerebellar cortex. Also, at least study finds some degree of cortical thickening.
Hearing loss. The other predominant feature of TBC1D24-related phenotypes is hearing loss. Mutations in TBC1D24 can cause autosomal recessive non-syndromic hearing loss (DFNB86) characterized by profound prelingual deafness. Autosomal dominant nonsyndromic hearing loss (DFNA65), also caused by TBC1D24 variants, is characterized by slowly progressive hearing loss affecting initially the high frequencies in the third decade.
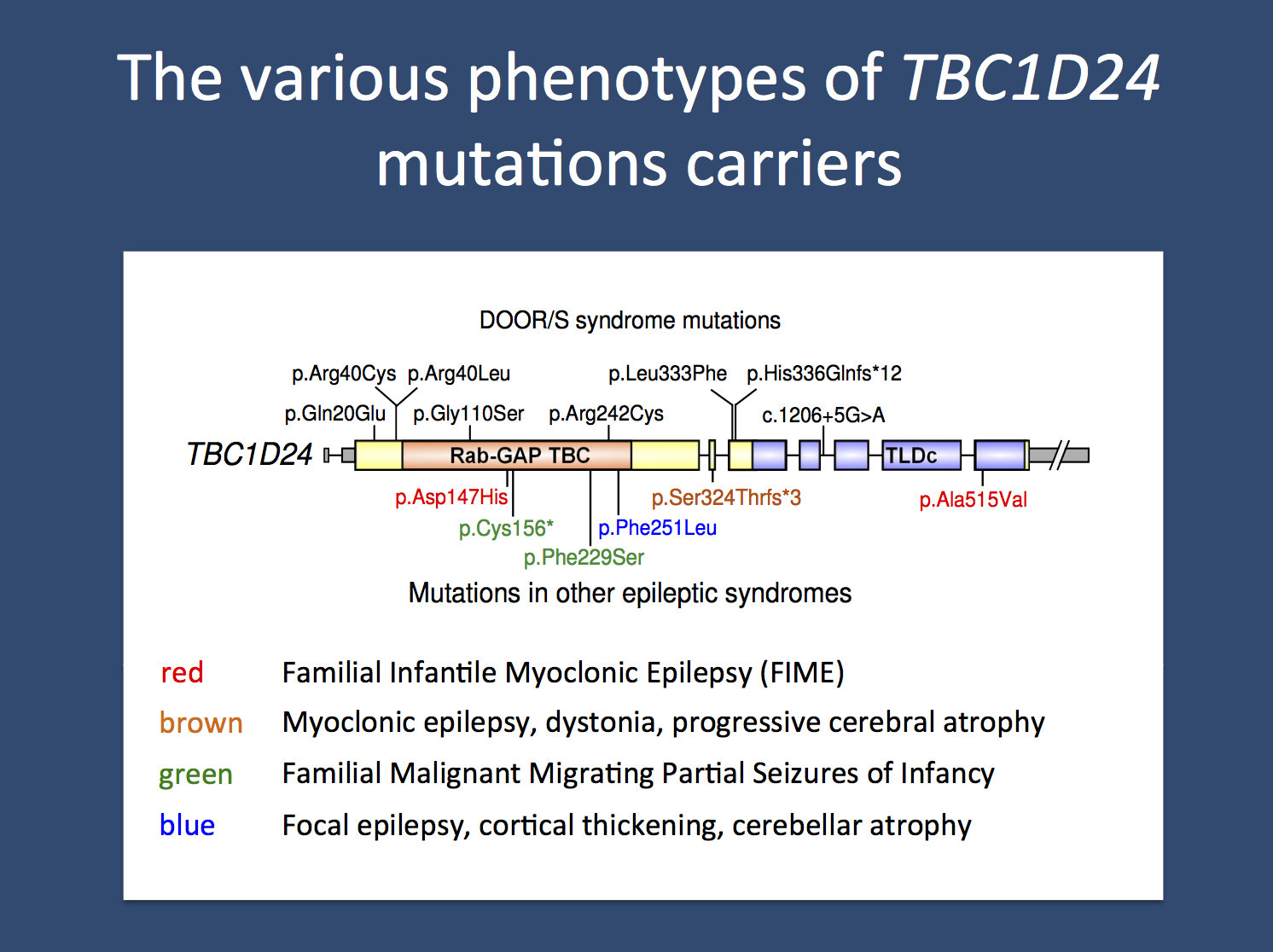
The various recessive and compound heterozygous epilepsy phenotypes due to mutations in TBC1D24.
Genetics
Mutation spectrum. Over 25 unique disease-causing variants have been reported in TBC1D24. The majority of these variants are missense, but a small number of nonsense, splice site, small deletions, and small insertions have also been identified. In addition, a single whole gene deletion was found in a patient with epilepsy with cognitive impairment and autism. Some of the variants are shown in the image above.
Genotype-Phenotype Correlation. There is currently no way to predict the epilepsy phenotype based on the genotype; A certain variant type (e.g., missense or nonsense) or region within the gene has not been associated with a specific epilepsy phenotype. However, no single variant has been reported to cause completely different phenotypes, such as isolated hearing loss versus DOORS syndrome A single missense variant (p.Ser178Leu) has been reported to cause autosomal dominant non-syndromic hearing loss in a small number of families, while a number of different missense variants (including p.Asp70Tyr, p.Arg214His, and p.Arg293Pro) have been reported to cause autosomal recessive, non-syndromic hearing loss .
General Considerations for Variant interpretation. When reviewing a genetic variant to determine its significance for a given patient, it is important to weigh multiple pieces of evidence:
Considerations for gene level interpretation. First, it is important to establish the strength of the evidence showing that the gene is associated with epilepsy. Some genes may only have one variant reported in a single individual with epilepsy, while other genes may have multiple variants reported in many large families with an autosomal dominant pattern of epilepsy. For TBC1D24, there is strong evidence for its role in human epilepsy.
Considerations for variant level interpretation. When reviewing the significance of a variant, it is important to consider the impact on the gene and the presence of the variant in previously described patient and control populations. Many clinical genetic testing laboratories classify genetic variants into different categories, ranging from benign to pathogenic. Variants that are common in control populations and would not be predicted to have a major impact on the gene/protein are generally classified as benign. Variants are more likely to be classified as pathogenic if the variants are rare or not present in the control population, reported in multiple individuals or families with disease, and likely to have a higher impact on the gene/protein based on the type of mutation or functional studies. Variants with uncertain or limited available evidence may be classified as variants of uncertain significance (VUS), indicating that further information is required in order for the variant to be further defined. In some cases, testing additional family members can be helpful, as it allows the lab to determine whether or not the variant was inherited (versus de novo) and how the variant segregates with disease in the family. Sometimes further classification of a VUS requires waiting for the identification of additional patients or families with similar or nearby variants.
Inheritance, Penetrance & Prevalence. Thus far, all TBC1D24-related epilepsy phenotypes are autosomal recessive and require biallelic disease-causing variants in order for an individual to be symptomatic. TBC1D24-related hearing loss has been reported to be caused by both heterozygous and biallelic variants with autosomal dominant and autosomal recessive inheritance, respectively. Penetrance of TBC1D24-related phenotypes appears to be high. Considerable variable expressivity can occur, particularly in seizure type/severity and in presence of non-seizure phenotypes. TBC1D24 variants and their associated phenotypes are rare, but the exact prevalence is unknown.
Mechanism & Function
Mechanism. TBC1D24 encodes a protein with two identifiable domains: a Tre2–Bub2–Cdc16 (TBC) domain similar to other RAB GTPase-activating proteins (RabGAPs), and a TLDc domain (TBC, LysM, Domain catalytic). The TBC/RabGAP domains interact with small GTPases, often helping to hydrolyze GTP and rendering the Rab proteins unable to interact with effectors, thus regulating the proper transport of intracellular vesicles. The function of the TLDc domain is not known but is thought to be involved in oxidative stress resistance. TBC1D24 has been demonstrated to interact with the ARF6 GTPase. In Drosophila, the ortholog Skywalker (Sky) facilitates endosomal trafficking in synaptic vesicles by facilitating GTP hydrolysis by Rab35, thus controlling synaptic vesicle rejuvenation and neurotransmitter release. Notably, mutating Sky caused synaptic vesicles to travel excessively to endosomes, so that older proteins are more efficiently degraded. These perturbations could affect proper regulation of neurotransmitter release and lead to epilepsy.
Cell/Animal models.
Mice. There is currently no mouse model for TBC1D24-related epilepsy disorders.
The Clinical Perspective
Recurrence risk & testing of family members. TBC1D24-related epilepsies are inherited in an autosomal recessive inheritance pattern. In order to inherit the condition, an individual must inherit two disease-causing TBC1D24 variants, one from each parent. Most children of an individual with TBC1D24-related epilepsy will be asymptomatic carriers, unless their other parent also carries a pathogenic TBC1D24 variant. Parents with a previous child with biallelic TBC1D24 variants generally have a 25% (1 in 4) chance that each future child will inherit both TBC1D24 variants. TBC1D24-related hearing loss can be inherited in an autosomal dominant or recessive inheritance pattern.
Therapy. There are no gene-specific treatments or specific contraindicated antiepileptic medications known for TBC1D24 at this time.
Community
Families with mutations in TBC1D24 can connect with other families using the links found here.
Research studies. The scientific community is actively studying TBC1D24and its role in human disease. The TBC1D24 Epilepsiome team is happy to facilitate if you have questions or a specific interest in this gene.
Family connections. The TBC1D24 community is active. More information is available on the webpage, including information about connecting to other families and becoming involved in research studies.
Resources & References
Websites
For Families: TBC1D24 Genetics Home Reference
For Clinicians: TBC1D24 GeneReview
Multiplex families with epilepsy: Success of clinical and molecular genetic characterization.
Afawi Z, Oliver KL, Kivity S, Mazarib A, Blatt I, Neufeld MY, Helbig KL, Goldberg-Stern H, Misk AJ, Straussberg R, Walid S, Mahajnah M, Lerman-Sagie T, Ben-Zeev B, Kahana E, Masalha R, Kramer U, Ekstein D, Shorer Z, Wallace RH, Mangelsdorf M, MacPherson JN, Carvill GL, Mefford HC, Jackson GD, Scheffer IE, Bahlo M, Gecz J, Heron SE, Corbett M, Mulley JC, Dibbens LM, Korczyn AD, Berkovic SF.
Neurology. 2016 Feb 23;86(8):713-22. doi: 10.1212/WNL.0000000000002404. Epub 2016 Jan 22.
PMID: 26802095
TBC1D24 mutation associated with focal epilepsy, cognitive impairment and a distinctive cerebro-cerebellar malformation.
Afawi Z, Mandelstam S, Korczyn AD, Kivity S, Walid S, Shalata A, Oliver KL, Corbett M, Gecz J, Berkovic SF, Jackson GD.
Epilepsy Res. 2013 Jul;105(1-2):240-4. doi: 10.1016/j.eplepsyres.2013.02.005. Epub 2013 Mar 19.
PMID: 23517570
Recessive TBC1D24 Mutations Are Frequent in Moroccan Non-Syndromic Hearing Loss Pedigrees.
Bakhchane A, Charif M, Salime S, Boulouiz R, Nahili H, Roky R, Lenaers G, Barakat A.
PLoS One. 2015 Sep 15;10(9):e0138072. doi: 10.1371/journal.pone.0138072. eCollection 2015.
PMID: 26371875
Phenotype-genotype complexities: opening DOORS.
Berkovic SF, Gecz J.
Lancet Neurol. 2014 Jan;13(1):24-5. doi: 10.1016/S1474-4422(13)70237-0. Epub 2013 Nov 29. No abstract available.
PMID: 24291219
DOORS syndrome: phenotype, genotype and comparison with Coffin-Siris syndrome.
Campeau PM, Hennekam RC; DOORS syndrome collaborative group.
Am J Med Genet C Semin Med Genet. 2014 Sep;166C(3):327-32. doi: 10.1002/ajmg.c.31412. Epub 2014 Aug 28.
PMID: 25169651
The genetic basis of DOORS syndrome: an exome-sequencing study.
Campeau PM, Kasperaviciute D, Lu JT, Burrage LC, Kim C, Hori M, Powell BR, Stewart F, Félix TM, van den Ende J, Wisniewska M, Kayserili H, Rump P, Nampoothiri S, Aftimos S, Mey A, Nair LD, Begleiter ML, De Bie I, Meenakshi G, Murray ML, Repetto GM, Golabi M, Blair E, Male A, Giuliano F, Kariminejad A, Newman WG, Bhaskar SS, Dickerson JE, Kerr B, Banka S, Giltay JC, Wieczorek D, Tostevin A, Wiszniewska J, Cheung SW, Hennekam RC, Gibbs RA, Lee BH, Sisodiya SM.
Lancet Neurol. 2014 Jan;13(1):44-58. doi: 10.1016/S1474-4422(13)70265-5. Epub 2013 Nov 29.
PMID: 24291220
A focal epilepsy and intellectual disability syndrome is due to a mutation in TBC1D24.
Corbett MA, Bahlo M, Jolly L, Afawi Z, Gardner AE, Oliver KL, Tan S, Coffey A, Mulley JC, Dibbens LM, Simri W, Shalata A, Kivity S, Jackson GD, Berkovic SF, Gecz J.
Am J Hum Genet. 2010 Sep 10;87(3):371-5. doi: 10.1016/j.ajhg.2010.08.001.
PMID: 20797691
Lack of pathogenic mutations in six patients with MMPSI.
De Filippo MR, Rizzo F, Marchese G, Giurato G, Nassa G, Ravo M, Tarallo R, Pironti E, Vecchi M, Crichiutti G, Capizzi G, Verrotti A, Weisz A, Coppola G.
Epilepsy Res. 2014 Feb;108(2):340-4. doi: 10.1016/j.eplepsyres.2013.11.007. Epub 2013 Nov 16.
PMID: 24315024
TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway.
Falace A, Buhler E, Fadda M, Watrin F, Lippiello P, Pallesi-Pocachard E, Baldelli P, Benfenati F, Zara F, Represa A, Fassio A, Cardoso C.
Proc Natl Acad Sci U S A. 2014 Feb 11;111(6):2337-42. doi: 10.1073/pnas.1316294111. Epub 2014 Jan 27.
PMID: 24469796
TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy.
Falace A, Filipello F, La Padula V, Vanni N, Madia F, De Pietri Tonelli D, de Falco FA, Striano P, Dagna Bricarelli F, Minetti C, Benfenati F, Fassio A, Zara F.
Am J Hum Genet. 2010 Sep 10;87(3):365-70. doi: 10.1016/j.ajhg.2010.07.020. Epub 2010 Aug 19.
PMID: 20727515
Reduced synaptic vesicle protein degradation at lysosomes curbs TBC1D24/sky-induced neurodegeneration.
Fernandes AC, Uytterhoeven V, Kuenen S, Wang YC, Slabbaert JR, Swerts J, Kasprowicz J, Aerts S, Verstreken P.
J Cell Biol. 2014 Nov 24;207(4):453-62. doi: 10.1083/jcb.201406026.
PMID: 25422373
TBC1D24 truncating mutation resulting in severe neurodegeneration.
Guven A, Tolun A.
J Med Genet. 2013 Mar;50(3):199-202. doi: 10.1136/jmedgenet-2012-101313. Epub 2013 Jan 23.
PMID: 23343562
Novel compound heterozygous mutations in TBC1D24 cause familial malignant migrating partial seizures of infancy.
Milh M, Falace A, Villeneuve N, Vanni N, Cacciagli P, Assereto S, Nabbout R, Benfenati F, Zara F, Chabrol B, Villard L, Fassio A.
Hum Mutat. 2013 Jun;34(6):869-72. doi: 10.1002/humu.22318. Epub 2013 Apr 12.
PMID: 23526554
TBC1D24-Related Disorders.
Mucha BE, Hennekam RCM, Sisodiya S, Campeau PM.
In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016.
2015 Feb 26.
PMID: 25719194
A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy.
Muona M, Berkovic SF, Dibbens LM, Oliver KL, Maljevic S, Bayly MA, Joensuu T, Canafoglia L, Franceschetti S, Michelucci R, Markkinen S, Heron SE, Hildebrand MS, Andermann E, Andermann F, Gambardella A, Tinuper P, Licchetta L, Scheffer IE, Criscuolo C, Filla A, Ferlazzo E, Ahmad J, Ahmad A, Baykan B, Said E, Topcu M, Riguzzi P, King MD, Ozkara C, Andrade DM, Engelsen BA, Crespel A, Lindenau M, Lohmann E, Saletti V, Massano J, Privitera M, Espay AJ, Kauffmann B, Duchowny M, Møller RS, Straussberg R, Afawi Z, Ben-Zeev B, Samocha KE, Daly MJ, Petrou S, Lerche H, Palotie A, Lehesjoki AE.
Nat Genet. 2015 Jan;47(1):39-46. doi: 10.1038/ng.3144. Epub 2014 Nov 17.
PMID: 25401298
Epilepsy genetics–past, present, and future.
Poduri A, Lowenstein D.
Curr Opin Genet Dev. 2011 Jun;21(3):325-32. doi: 10.1016/j.gde.2011.01.005. Epub 2011 Jan 27. Review.
PMID: 21277190
Homozygous TBC1D24 mutation in two siblings with familial infantile myoclonic epilepsy (FIME) and moderate intellectual disability.
Poulat AL, Ville D, de Bellescize J, André-Obadia N, Cacciagli P, Milh M, Villard L, Lesca G.
Epilepsy Res. 2015 Mar;111:72-7. doi: 10.1016/j.eplepsyres.2015.01.008. Epub 2015 Jan 25.
PMID: 25769375
Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86.
Rehman AU, Santos-Cortez RL, Morell RJ, Drummond MC, Ito T, Lee K, Khan AA, Basra MA, Wasif N, Ayub M, Ali RA, Raza SI; University of Washington Center for Mendelian Genomics, Nickerson DA, Shendure J, Bamshad M, Riazuddin S, Billington N, Khan SN, Friedman PL, Griffith AJ, Ahmad W, Riazuddin S, Leal SM, Friedman TB.
Am J Hum Genet. 2014 Jan 2;94(1):144-52. doi: 10.1016/j.ajhg.2013.12.004.
PMID: 24387994
Early-onset epileptic encephalopathy with hearing loss in two siblings with TBC1D24 recessive mutations.
Stražišar BG, Neubauer D, Paro Panjan D, Writzl K.
Eur J Paediatr Neurol. 2015 Mar;19(2):251-6. doi: 10.1016/j.ejpn.2014.12.011. Epub 2014 Dec 20.
PMID: 25557349