
KCNT1. This is the Epilepsiome page on KCNT1, a gene primarily associated with two distinct genetic epilepsy syndromes, namely Migrating Partial Seizures of Infancy (MPSI) and Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (ADNFLE). Ever since its discovery in 2012, KCNT1 mutations are increasingly recognized in severe, early onset epilepsies. Some studies suggest that precision medicine with quinidine may help reduce seizure burden in some cases.
Here are the most recent blog posts on KCNT1
- This is what you should know about KCNT1 – a 2015 update
- A new spectrum unfolding – KCNT1 mutations in ADNFLE and MMPSI
- This is what you will see in epilepsy genetics in the next five years
- Five unexpected things I realized at AES this year
In a nutshell. KCNT1 is not getting any less mysterious. KCNT1 variants were originally described as causing a very rare, but distinct catastrophic epilepsy of childhood: migrating partial seizures of infancy (MPSI). The very same variants were also discovered to result in a severe autosomal dominant form of frontal lobe epilepsy with prominent psychiatric features (ADNFLE). In addition, KCNT1 variants have been identified in patients presenting with other early-onset epileptic encepholopathies, including West syndrome and Ohtahara syndrome, and focal epilepsies, as well as leukodystrophy and leukoencephalopathy with severe epilepsy. Several mutation hotspots and recurrent variants in KCNT1 have emerged, most prominently the clustering around the functional domains of the intracellular C-terminus, particularly the RCK domain and the NAD+ binding domain; however, the exact same variants can cause different phenotypes, indicating that the genotype-phenotype correlation is not clear-cut.
| Phenotypes | Genetics | Mechanism & Function |
| The Clinical Perspective | Community | Resources & References |
Phenotypes
MPSI. Migrating partial seizures of infancy is one of the two main epilepsy syndromes connected to KCNT1. (MPSI is also referred to as epilepsy of infancy with migrating focal seizures, migrating partial epilepsy of infancy, and malignant migrating focal seizures of infancy.) MPSI is a severe epilepsy syndrome that starts in the first six months of life. This severe epileptic encephalopathy is characterized by a wide spectrum of focal seizures and arrest of psychomotor development. On EEG, seizures can be observed to arise almost randomly from various regions of the brain. Seizures may migrate from one brain region to another, which is an extremely unusual feature that has given MPSI its name. Initial MRI images are normal, and except for two patients with mutations in SCN1A, the cause of MPSI was unknown prior to the discovery of KCNT1. MPSI is rare with fewer than 100 patients described worldwide. The first pathogenic KCNT1 variants were identified in patients with MPSI by Barcia and colleagues in 2012. It has become apparent that KCNT1 variants are the most common cause of MPSI, accounting for almost 40% of patients (Lim et al, 2016).

Figure. Distribution of the KCNT1 mutations along the protein, demonstrating the clustering of mutations around the functional domains of the C-terminus. On the left side, a pedigree is shown where affected individuals have either nocturnal frontal lobe seizures or Migrating Partial Seizures of Infancy (MPSI or MMFSI). (non-copyrighted version of the figures provided by the author).
ADNFLE. Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (ADNFLE) is a familial epilepsy syndrome, which is characterized by nocturnal frontal lobe seizures. Nocturnal frontal lobe seizures are very distinct events that are characterized by nocturnal arousal, screaming, violent movement of the legs (bipedal automatisms) and sometimes the feeling of choking. These events are often difficult to distinguish from sleep disorders (parasomnias). The genetic basis of ADNFLE is the very start of modern epilepsy genetics. As the first epilepsy gene, a mutation in CHRNA4 was reported in a large Australian family with ADNFLE in 1995. Further families were found to have mutations in CHRNB2 and CHRNA2. All three genes code for nicotinergic acetylcholine receptors, which appeared to be the unifying molecular genetic theme for ADNFLE until the report by Heron and collaborators in 2012. Several families had been reported in the literature, where nocturnal frontal lobe seizures are accompanied by psychiatric features and intellectual disability. Most of these families were found to be negative for pathogenic variants in the three previously known genes. Using exome sequencing, Heron and collaborators identified disease-causing variants in KCNT1 in an Australian family with six affected individuals. KCNT1 variants were also found in three additional smaller families. In most patients the epilepsy was more severe than in patients with pathogenic variants in genes for nicotinergic receptors and there was a higher frequency of intellectual disability and psychiatric problems, including depression, anxiety, and attention-deficit hyperactivity disorder. Moller and collaborators described additional families with KCNT1 variants.
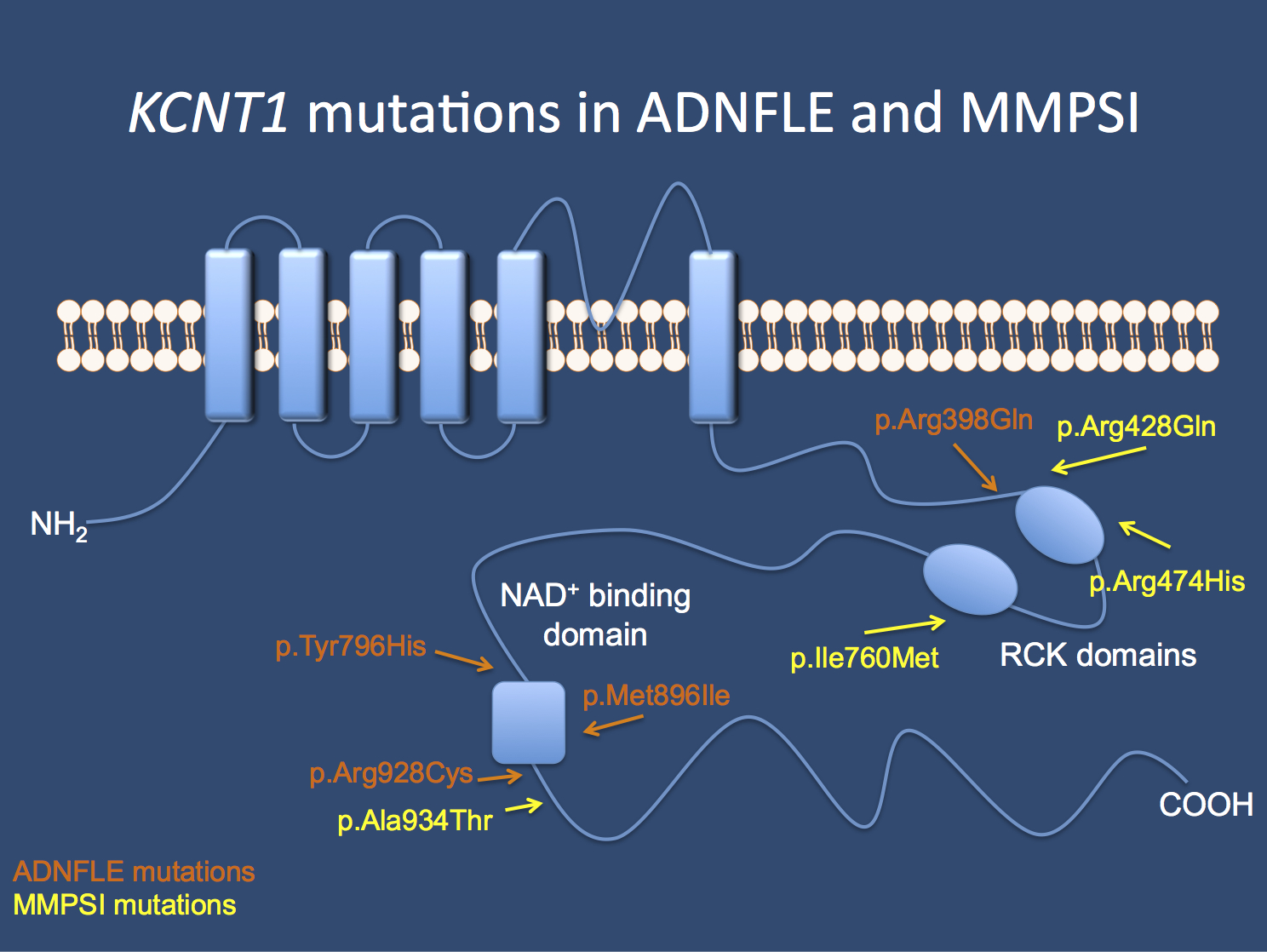
The KCNT1 channel. Mutations in the gene cause Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (ADNFLE) and Malignant Migrating Partial Seizures of Infancy (MMPSI) as identified in the initial publication by Barcia and Heron.
Other Phenotypes. Heterozygous KCNT1 variants have also been identified in patients with a clinical diagnosis of other early onset epileptic encephalopathies and focal epilepsies, including West syndrome, leukoencephalopathy with severe epilepsy, multifocal epilepsy and hypomyelinating leukodystrophy. In addition, a single patient with Ohtahara syndrome was identified to have a homozygous variant in KCNT1 as a result of uniparental disomy.
Cardiac arrhythmias. Several patients with KCNT1 variants have had cardiac arrhythmias, suggesting a possible association between KCNT1 and cardiac disorders. KCNT1 is expressed in the heart. Further studies are needed to confirm this association.
Genetics
Mutation Spectrum (Type and Location). Thus far, over 20 different disease-causing heterozygous missense variants have been reported in KCNT1 (Lim et al, 2016). One variant (p.Ala966Thr) in a patient with Ohtahara syndrome was identified in the homozygous state due to uniparental isodisomy. Several family members who were heterozygous for this variant had a history of childhood idiopathic epilepsy, while at least one individual was reportedly asymptomatic (Martin et al, 2014), suggesting that this variant is associated with a form of autosomal dominant epilepsy with incomplete penetrance but causes a more severe phenotype in the homozygous state. Most variants associated with an epilepsy phenotype cluster around specific functional domains of the long intracellular C terminus, suggesting that regulation of this ion channel may be an important mechanism to target for therapy development. Multiple recurrent variants have been identified (Table 1 below), indicating that mutational hot spots exist in KCNT1.
Table 1. Recurrent mutations in KCNT1
| Mutation | Phenotype | Frequency |
| p.Gly288Ser | ADNFLE, MMFSI | 6x |
| p.Arg398Gln | ADNFLE, MMFSI | 3x |
| p.Arg428Gln | MMFSI | 4x |
| p.Arg928Cys | ADNFLE | 3x |
| p.Ala934Thr | MMFSI | 3x |
Genotype-Phenotype Correlation. No clear genotype-phenotype correlations have been reported, though the number of patients described is still small. As shown in Table 1 above, even recurrent variants can cause different clinical presentations. The same variant can cause MPSI, ADNFLE and hypomyelinating leukodystrophy, even within the same family. This phenotypic variation suggests that there are modifying factors, such as environmental effects or modifier genes, that impact the resulting clinical features.
General Considerations for Variant interpretation. When reviewing a genetic variant to determine its significance for a given patient, it is important to weigh multiple pieces of evidence:
Gene level interpretation. First, it is important to establish the strength of the evidence showing that the gene is associated with epilepsy. Some genes may only have one variant reported in a single individual with epilepsy, while other genes may have multiple variants reported in many large families with an autosomal dominant pattern of epilepsy.
Variant level interpretation. When reviewing the significance of a variant, it is important to consider the impact on the gene and the presence of the variant in previously described patient and control populations. Many clinical genetic testing laboratories classify genetic variants into different categories, ranging from benign to pathogenic. Variants that are common in control populations and would not be predicted to have a major impact on the gene/protein are generally classified as benign. Variants are more likely to be classified as pathogenic if the variants are rare or not present in the control population, reported in multiple individuals or families with disease, and likely to have a higher impact on the gene/protein based on the type of mutation or functional studies. Variants with uncertain or limited available evidence may be classified as variants of uncertain significance (VUS), indicating that further information is required in order for the variant to be further defined. In some cases, testing additional family members can be helpful, as it allows the lab to determine whether or not the variant was inherited (versus de novo) and how the variant segregates with disease in the family. Sometimes further classification of a VUS requires waiting for the identification of additional patients or families with similar or nearby variants.
Inheritance, Penetrance & Prevalence. KCNT1 variants can be de novo or inherited in an autosomal dominant pattern. Penetrance of the phenotype appears to be high, but multiple individuals carrying familial KCNT1 variants have been reported to be seizure free. Considerable variable expressivity, particularly in seizure type and severity, can occur, even with the same variant within the same family. KCNT1 variants and their associated phenotypes are rare, but the exact prevalence is unknown. According to one study, KCNT1 variants were identified in approximately 3% of patients with early-onset epileptic encephalopathies, with the greatest percentage of patients with KCNT1 variants having a diagnosis of MPSI.
Mechanism & Function
SLICK and SLACK. KCNT1 and KCNT2 are sodium-activated potassium channels; they are permeable for potassium ions in response to sodium. Both channels are highly expressed in the nervous system and contribute to the slow hyperpolarisation after repetitive neuronal firing. Both ion channels are also referred to as SLACK (KCNT1) and SLICK (KCNT2), which emphasize their role in the SLOW part of the hyperpolarisation. The action of potassium channels is needed to return the membrane potential to the resting membrane potential after neuronal firing.
With more than 80 members, the family of human potassium channels is highly diverse, and probably many different potassium channels contribute to the overall potassium current during different stages of development. For example, KCNQ2 and KCNQ3 likely play an important role in the first months of life, and, therefore, pathogenic variants in these genes cause benign familial neonatal seizures (BFNS). While the precise role of KCNT1 is not yet known, some of the most recent findings are puzzling. The MPSI variants identified by Barcia and collaborators appear to be activating, i.e. increasing the potassium current. How an increase in potassium current translates into hyperexcitability is difficult to explain. The increased potassium current would drive the neuronal membrane potential towards hyperpolarisation, thereby decreasing excitability. However, the authors also indicate that KCNT1 is highly expressed during neuronal development and may also have additional functions.
Cell/Animal models.
Mice. The KCNT1 null mouse presents with slightly reduced locomotive activity and impairment of both reverse learning memory and adaptation to new environments. The mice do not have seizures (Bausch et al, 2015).
The Clinical Perspective
Recurrence Risk. KCNT1 variants can be de novo or inherited in an autosomal dominant pattern. At least two pathogenic variants were found to be inherited from unaffected parents with somatic mosaicism, which is important for recurrence risk counseling. Although not yet reported, germline mosaicism of KCNT1 variants is possible, making the recurrence risk for families with one child with a KCNT1 variant higher than the general population risk.
Therapy
Quinidine. The old antiarrhythmic drug quinidine was found to inhibit the KCNT1 channel, counteracting the gain-of-function that was introduced by the mutation. In 2014, a functional and aclinical publication could demonstrate that quinidine can be used for targeted treatment of MPSI due to activating KCNT1 mutations, providing us with a successful example of precision medicine in epilepsy genetics. While larger studies on the effect of quinidine in KCNT1-mediated seizures are still in process, the quinidine story is a nice example of how genetic discovery can point towards existing drugs that can be used. Quinidine has been around for ages, but there was never any impetus to try it for seizures. Quinidine is not a proven treatment for MPSI and further studies are planned to fully evaluate its potential use in patients with this diagnosis.
Resources & References
Websites
For Families:
KCNT1 Genetics Home Reference – https://ghr.nlm.nih.gov/gene/KCNT1
For Clinicians:
GeneReviews: Autosomal Dominant Nocturnal Frontal Lobe Epilepsy
http://www.ncbi.nlm.nih.gov/books/NBK1169/
References
Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, Chen H, Kronengold J, Abhyankar A, Cilio R, Nitschke P, Kaminska A, Boddaert N, Casanova JL, Desguerre I, Munnich A, Dulac O, Kaczmarek LK, Colleaux L, Nabbout R. 2012. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. 44(11):1255-9. PMID: 23086397
Bausch AE, Dieter R, Nann Y, Hausmann M, Meyerdierks N, Kaczmarek LK, Ruth P, Lukowski R. 2015. The sodium-activated potassium channel Slack is required for optimal cognitive flexibility in mice. Learn Mem. 15;22(7):323-35. PMID: 26077685
Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. 2014. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann Neurol. 76(3):457-61. PMID:25042079
Boillot M, Baulac S. 2016. Genetic models of focal epilepsies. J Neurosci Methods. 15;260:132-43. PMID: 26072248
Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, Oliver KL, Mazarib A, Afawi Z, Korczyn A, Plazzi G, Petrou S, Berkovic SF, Scheffer IE, Dibbens LM. 2012. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 44(11):1188-90. PMID: 23086396
Ishii A, Shioda M, Okumura A, Kidokoro H, Sakauchi M, Shimada S, Shimizu T, Osawa M, Hirose S, Yamamoto T. 2013. A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene. 531(2):467-71. PMID: 24029078
Lim CX, Ricos MG, Dibbens LM, Heron SE. 2016. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects. J Med Genet. [Epub ahead of print] PMID: 26740507
Martin HC, Kim GE, Pagnamenta AT, Murakami Y, Carvill GL, Meyer E, Copley RR, Rimmer A, Barcia G, Fleming MR, Kronengold J, Brown MR, Hudspith KA, Broxholme J, Kanapin A, Cazier JB, Kinoshita T, Nabbout R; WGS500 Consortium, Bentley D, McVean G, Heavin S, Zaiwalla Z, McShane T, Mefford HC, Shears D, Stewart H, Kurian MA, Scheffer IE, Blair E, Donnelly P, Kaczmarek LK, Taylor JC. 2014. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet. 23(12):3200-11. PMID: 24463883
McTague A, Appleton R, Avula S, Cross JH, King MD, Jacques TS, Bhate S, Cronin A, Curran A, Desurkar A, Farrell MA, Hughes E, Jefferson R, Lascelles K, Livingston J, Meyer E, McLellan A, Poduri A, Scheffer IE, Spinty S, Kurian MA, Kneen R. 2013. Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum. Brain. 136(Pt 5):1578-91. PMID: 23599387
Mikati MA, Jiang YH, Carboni M, Shashi V, Petrovski S, Spillmann R, Milligan CJ, Li M, Grefe A, McConkie A, Berkovic S, Scheffer I, Mullen S, Bonner M, Petrou S, Goldstein D. 2015. Quinidine in the treatment of KCNT1-positive epilepsies. Ann Neurol. 78(6):995-9. PMID: 26369628
Milligan CJ, Li M, Gazina EV, Heron SE, Nair U, Trager C, Reid CA, Venkat A, Younkin DP, Dlugos DJ, Petrovski S, Goldstein DB, Dibbens LM, Scheffer IE, Berkovic SF, Petrou S. 2014 KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol. Apr;75(4):581-90. PMID: 24591078
Ohba C, Kato M, Takahashi N, Osaka H, Shiihara T, Tohyama J, Nabatame S, Azuma J, Fujii Y, Hara M, Tsurusawa R, Inoue T, Ogata R, Watanabe Y, Togashi N, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Tanaka F, Saitsu H, Matsumoto N. 2015. De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia. Sep;56(9):e121-8. PMID: 26140313
Tang QY, Zhang FF, Xu J, Wang R, Chen J, Logothetis DE, Zhang Z. 2016. Epilepsy-Related Slack Channel Mutants Lead to Channel Over-Activity by Two Different Mechanisms. Cell Rep. 14(1):129-39. PMID: 26725113
Vanderver A, Simons C, Schmidt JL, Pearl PL, Bloom M, Lavenstein B, Miller D, Grimmond SM, Taft RJ. 2014. Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr Neurol. Jan;50(1):112-4. PMID: 24120652