
STX. I have to admit that the main gene that our team is working on has received relatively little attention on our blog, even as several manuscripts on STXBP1 have recently been published that add to our understanding of STXBP1-related disorders. I described STXBP1 at the Simons Foundation’s INSYNC-AS meeting as one of the genes with the “fastest growing knowledge,” now following close behind recognized neurodevelopmental disorders including Rett and Angelman Syndrome. In this post, I would like to feature one of our recent publications in Neurology Genetics that assessed the developmental trajectory of 48 individuals with STXBP1-related disorders. In particular, I would like to zero in on the most important result of this study, which expands on one of the controversies in the STX field.
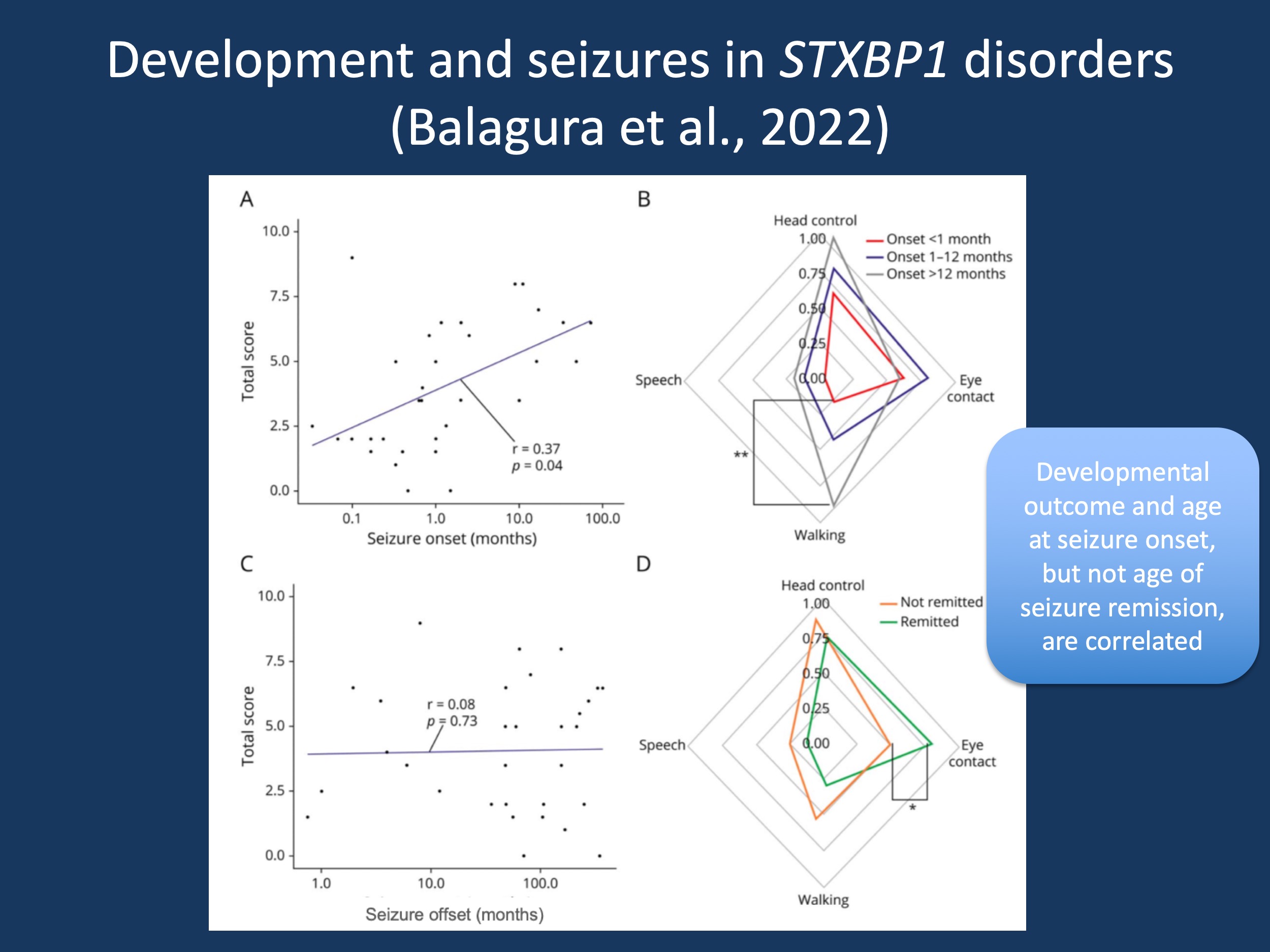
Figure 1. (A). STXBP1_DevScore distribution in n = 36 individuals with STXBP1-DEE ≥3 years old (median age 8.35 years, range 3–35 years) based on age at seizure onset (log10 scale). Seizure onset <1 month (n = 15) 1–12 months (n = 16), >12 months (n = 5). (Pearson correlation coefficient). (B) Developmental milestones subscores (mean) stratified per age range at seizure onset. **p = 0.0049, one-way ANOVA. (C) STXBP1_DevScore distribution based on age at seizure offset or age at last examination in the case of active epilepsy (log10 scale). Active epilepsy (n = 23), seizure free (n = 13) (Pearson correlation coefficient). (D) Developmental milestones subscores (means) stratified per epilepsy outcomes. Active epilepsy (n = 23), seizure free (n = 13). *p = 0.0348. Source: Figure 3 in Balagura et al., 2022, reproduced under a Creative Commons Attribution-NonCommercial-NoDerivatives License 4.0 (CC BY-NC-ND).
STXBP1. For our readers who are not familiar with STXBP1, this gene codes for the MUNC18 protein, which is critical for synaptic vesicle fusion. The gene was initially identified in human disease in 2008 in a cohort of individuals with Ohtahara Syndrome by Saitsu and collaborators. However, soon after the initial discovery, the phenotypic range broadened to constitute a wide range of phenotypes, including intellectual disability without seizures, autism, infantile spasms, tremor, and movement disorders. While the phenotypic range in many genetic epilepsies is quite wide, it is particularly striking in STXBP1-related disorders.
Development and seizures. Individuals with the same variant can have different phenotypes and the association between specific variants and clinical features is not prominent, even though some significant findings exist. Moreover, in our initial publication by Stamberger and collaborators, we stipulated that the degree of intellectual disability and epilepsy are not correlated. This particular aspect is now addressed in detail in the publication by Balagura and collaborators, resulting in a surprising finding.
Onset. One of the main messages of the study by Balagura and collaborators is that there is a correlation between age of onset and developmental outcome. Our study used an STXBP1-scoring system, developed by international expert consensus, to compare disease severity across individuals. The system included analysis of a child’s neurological exam across childhood, as well as ability to make eye contact with other people, to meet motor milestones, and to use speech to communicate. The number of milestones achieved was correlated with seizure onset. This observation is critical as it adds to our initial discussion whether severity in STXBP1 can be measured within a single dimension. While we were initially skeptical in our publication by Stamberger and collaborators, our more recent study may suggest that the concept of measuring overall disease severity in STXBP1 is meaningful.
Offset. However, there was no observed correlation between developmental outcomes (milestones achieved) and age at seizure offset. This information informs clinical practice. For example, when we compare individuals with STXBP1-related disorders and infantile spasms starting between 3 months and 6 months, some individuals have immediate cessation of seizures with the first medication, while other individuals may require several medications with ongoing seizures into the second year of life. The lack of correlation of outcomes with age of offset suggests that the seizure trajectory in these cases has relatively little impact on developmental outcomes, even though the size of our study may have missed a smaller effect. While this is based on small numbers, this is an intriguing hypothesis to follow: ongoing seizures in STXBP1-related disorder may have less of an impact on development than we would usually think.
Further thoughts. In this post, I am only able to review a few of the results from our study, which is an Open Access publication. Here are two different aspects. First, the study included a monozygotic twin pair with a protein-truncating variant. Both twins had largely similar phenotypes. When contrasted with the wide range of phenotypes seen for individual variants between individuals, this may suggest that genetic background, potentially measurable through polygenic risk scores, may account for much of the variability. Second, the study by Balagura and collaborators reports on a second individual with STXBP1-related disorder and Focal Cortical Dysplasia (FCD) who experienced significant seizure reduction following epilepsy surgery. This reinforces the notion that, in contrast to Dravet Syndrome, a presurgical work-up in STXBP1 is meaningful – an STXBP1 diagnosis should not prevent consideration for epilepsy surgery.
This is what you have to know. In our study by Balagura and collaborators, we assessed the correlation between epilepsy features and developmental aspects. We found a correlation between developmental outcomes and seizure onset, demonstrating that severity and outcomes in STXBP1-related disorders can potentially be assessed through a single method rather than thinking about seizure severity and developmental outcome as two independent outcomes. However, we did not find a correlation of developmental outcome with the age of seizure remission, suggesting that longer seizure may not necessarily result in differences in developmental outcomes. In addition, the cohort analyzed by the paper may have been weighted towards individuals with STXBP1 who have severe epilepsy presentations, as 33% of individuals included had burst-suppression present on their EEG at one point, consistent with an Ohtahara syndrome presentation. Overall, the study by Balagura and collaborators gives us meaningful directions that future detailed natural history studies can focus on.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
