
Between the ion channels. Rather than going “beyond the ion channel,” in this post, we aim to look between them. We want to dive into a study where examining the group of epilepsy-related sodium channels was initially more informative than the single gene itself—even when that gene was SCN1A, the most established epilepsy gene. A recurrent SCN1A variant turned out to be part of an emerging, previously underappreciated gain-of-function spectrum. Here, we discuss the unusual phenotype of SCN1A gain-of-function variants and how we are currently working on integrating information on paralogs into the official ACMG variant curation criteria.
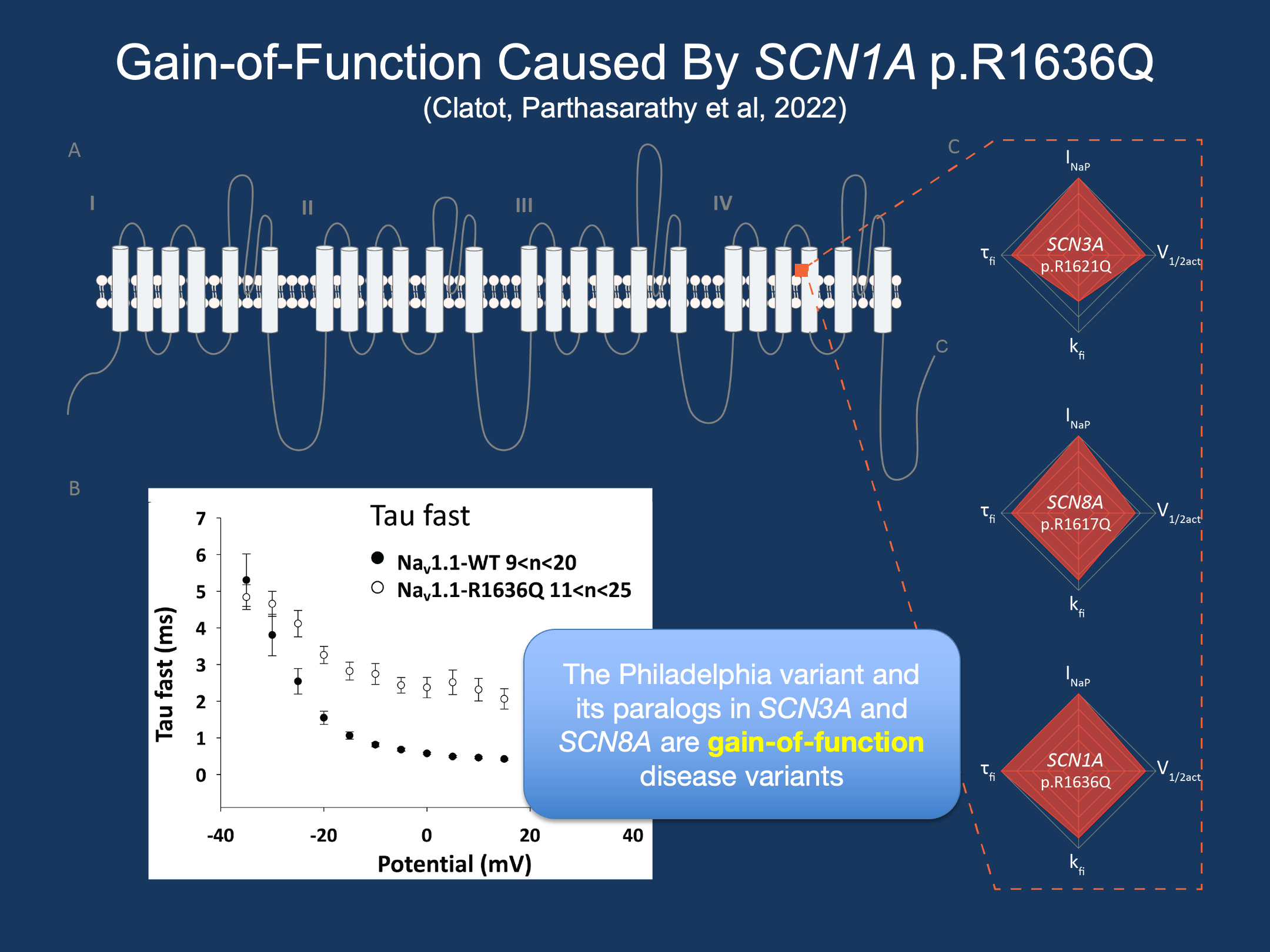
Figure 1. (A) The epilepsy sodium channels SCN1A, SCN2A, SCN3A, and SCN8A, have a common structure (B) The SCN1A Philadelphia variant, p.R1636Q, exhibits slower channel inactivation, a gain-of-function effect. (C) Identical, known gain-of-function variants in SCN3A and SCN8A have similar electrophysiological changes (adapted from Clatot, Parthasarathy et al., 2022, Epilepsia)
Para-logic. The sodium channel genes SCN1A through SCN11A are paralogs, originating from the same sequence and thus retaining incredibly high sequence similarity. Of these, SCN1A, SCN2A, SCN3A, and SCN8A are expressed in the brain and associated with epilepsy. These four genes have particularly high homology, which is very distinct within the larger gene family. The protein structures are nearly identical, featuring four domains with six transmembrane segments. Functional elements, such as the inactivation gate between DIII and DIV, the voltage-sensing function of S4, and the pore loop formed by S5-S6, are also preserved between channels. Even more granularly, individual amino acid residues can be mapped to one another, such as the arginine at position 853 in SCN2A, which matches that at position 862 in SCN1A and 847 in SCN8A.
The Philadelphia Variant. Loss-of-function variants in SCN1A cause Dravet syndrome, by far the most well-established gene-disease relationship in the epilepsies. However, over the course of two years, four individuals with the same de novo missense variant in SCN1A, p.R1636Q, presented to our clinic with disorders that were very different from Dravet syndrome. All individuals had seizures beginning in the first two months of life, far earlier than should be possible in Dravet syndrome given the dynamic transcription pattern of SCN1A. It is the most common SCN1A variant seen at our clinic, yet the corresponding presentations were completely different from the most common SCN1A-related epilepsy. What was different about this variant?
EMR, extrapolation, electrophysiology. Our lab has pioneered approaches to using information already within the electronic medical record (EMR) to trace clinical trajectories over time. We applied this idea using an automated, EMR-based proxy for age of seizure onset to assess seizure histories of individuals with SCN1A-related disorders. Individuals with the Philadelphia variant ranked in the earliest 5%. In addition, all four individuals had a seizure pattern distinct from those with known Dravet syndrome. Even more intriguing, identical substitutions to p.R1636Q in SCN2A (p.R1626Q), SCN3A (p.R1621Q), and SCN8A (p.R1617Q) were previously reported to cause disorders associated with gain-of-function in the respective channels. Electrophysiological studies in SCN3A and SCN8A had shown gain-of-function effects at the level of the ion channel as well. From this combination of clinical and functional data across the gene family, we hypothesized that SCN1A p.R1636Q was also a gain-of-function variant, the opposite mechanism of what can cause Dravet syndrome.
Gain-of-function in SCN1A. In our study by Clatot et al., we assessed the functional changes caused by the Philadelphia variant through electrophysiological assays. Typically, loss-of-function variants causing Dravet syndrome exhibit a strong reduction in the maximum amount of current conducted by the channel. Analysis of this variant revealed that the maximum current was not different from typically functioning SCN1A channels. Instead, channels tend to inactivate later and slower, meaning more channels remain open for longer after becoming activated. These properties are among the numerous, heterogeneous features that are implicated in gain-of-function of sodium channels—confirming that, in summary, SCN1A p.R1636Q is a gain-of-function variant in SCN1A.
Sodium channel blockers. Typically, when we receive genetic testing results with diagnostic SCN1A variant, one of the first things we think about is appropriate anti-seizure medication (ASM) use. The epilepsy community has diligently worked to spread the knowledge about sodium channel blockers being contraindicated in individuals with Dravet syndrome. However, the use of sodium channel blockers may be the treatment of choice for individuals with gain-of-function variants in SCN1A. Indeed, one individual in our cohort responded to oxcarbazepine. Functional assessment of the Philadelphia variant corroborated the efficacy of oxcarbazepine, showing that administration of this medication partially corrects the gain-of-function effects of the variant.
Variant curation. The Philadelphia variant story represents an example of the utility of synthesizing data across genes. For the epilepsy sodium channels, the idea of similarity across genes is finding its way into clinical variant interpretation. Clinical variant interpretation is underlain by the ACMG rules, which define what evidence matters and how much you need in order to confidently claim that a variant is pathogenic or benign. Within the ClinGen consortium, Variant Curation Expert Panels (VCEP) are created in order to modify these rules to be well-tailored to specific sets of genes. Within the ClinGen world, we are currently tasked with modifying variant interpretation criteria for epilepsy-related sodium channels within the framework of the Epilepsy Sodium Channel VCEP. In our draft specifications, we show that more than one identical, pathogenic substitutions in a different sodium channel increases the likelihood of pathogenicity nearly 10-fold. In other words, if a novel variant in SCN2A is identified, knowledge of pathogenic variants in the corresponding sites in SCN1A and SCN8A is more informative than the variant being confirmed de novo.
What you need to know. The Philadelphia variant, p.R1636Q, is the most common reported gain-of-function variant in SCN1A with a distinct phenotypic and functional profile. The emerging story on gain-of-function variants in SCN1A may give clinicians pause when considering the use of sodium channel blockers in epilepsy treatment when the individual’s phenotype is not consistent with the clinical entity of Dravet syndrome. Moreover, increasingly, as with our initial recognition of the gain-of-function effect of this variant, we are seeing that the specific sodium channel gene matters less than we might expect. Of course, each gene has its own phenotypic spectrum, but the homology between channels is so strong that a pathogenic variant in one gene can tell us just as much as a pathogenic variant in another gene. In dealing with the exponential growth of variants of uncertain significance, a surprisingly large amount of information is already known about disease variants if we look past the individual gene.
This blog post was co-written by Shridhar Parthasarathy and Stacey Cohen.
Shridhar Parthasarathy is a Data Scientist in the Helbig Lab, Children’s Hospital of Philadelphia.
