
Unravelling the BAFME mystery. The mystery surrounding Benign Adult Familial Myoclonic Epilepsy (BAFME) – also known as Familial Adult Myoclonic Epilepsy (FAME) or Familial Cortical Myoclonic Tremor and Epilepsy (FCMTE) – has persisted for years. BAFME is an autosomal dominant neurological disorder characterized by adult onset of myoclonic/cortical tremor and infrequent seizures. The clinical course is typically considered to be benign. Linkage studies have shown linkage to several regions including 8q24, 2p11.1-q12.2, 3q26.32-q28, and 5p15. A recent publication identified a variant in CTNND2 segregating with disease in a Dutch family with BAFME3, although it remains to be determined how broadly applicable CTNND2 variants are in other individuals with BAFME. Now in an elegant set of experiments by Ishiura and colleagues, a significant proportion of BAFME appears to be solved and is due to expansions of pentanucleotide intronic sequences in SAMD12.
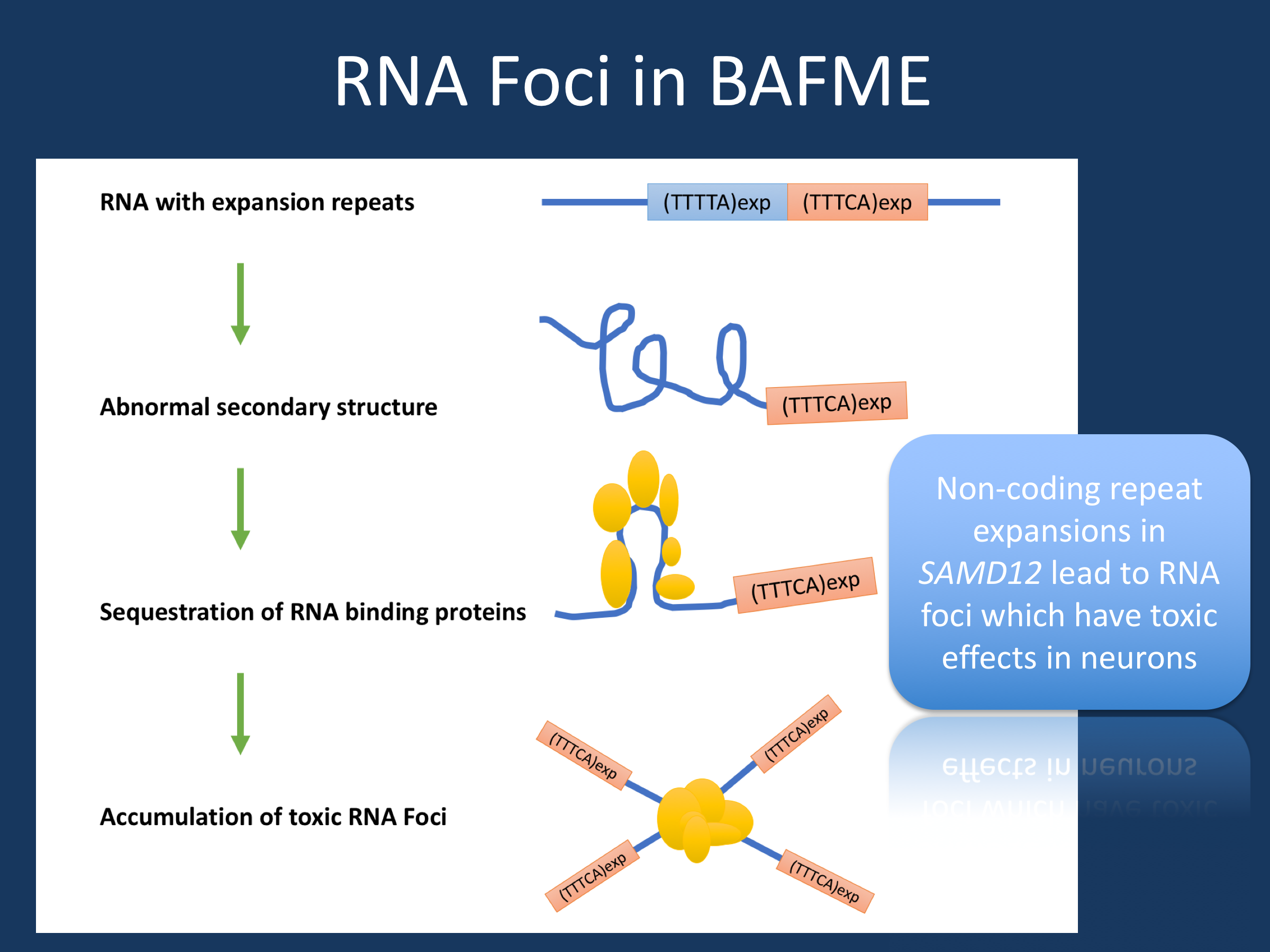
Accumulation of RNA Foci in BAFME. A simplified schematic diagram of how RNA foci accumulate in patients with TTTTA and TTTCA repeat expansions. The RNA contains an abnormal number of repeat expansions. This RNA then takes on an abnormal secondary structure. This abnormal RNA then recruits and sequesters RNA binding proteins. These clusters form aggregates called RNA foci that accumulate within the nuclei in cortical neurons and Purkinje cells. These RNA foci have toxic effects within the cell. RNA foci only containing TTTCA repeats have been observed in autopsied brain tissue of patients with BAFME.
A surprising repeat expansion disorder. Ishiura and colleagues identified expansions of non-coding TTTCA- and TTTTA-sequences in an intron of SAMD12 in 49/51 (96%) Japanese families with BAFME1, which links to chromosomal region 8q24. The repeat sizes were highly variable, ranging from 440 to 3680 repeats. Somewhat unsurprisingly, larger expansion sizes were correlated with earlier onset of seizures and myoclonic tremor. This is similar to what we observe in other repeat expansion disorders including Huntington Disease, although curiously not Unverricht-Lundborg Disease, another repeat expansion-associated myoclonic epilepsy. Four individuals homozygous for the SAMD12 repeat expansion were observed in the cohort, and these patients experienced younger age of onset and more severe disease progression, including impairment in walking and cognitive decline, which was not observed among heterozygotes. Two families were not found to have SAMD12 intronic pentanucleotide repeat expansions. However, affected individuals from these two families were found to have TTTTA and TTTCA repeat expansions in two other genes: one family had expansions in TNRC6A and one in RAPGEF2. TTTTA expansions in all three genes were identified in a small percentage (5% or less) of healthy controls; however no control individuals were found to have TTTCA expansions in SAMD12, TNRC6A, or RAPGEF2, suggesting that TTTCA repeat expansions may play a primary role in disease etiology.
What does SAMD12 do? The answer is: it may not actually matter. Little is known about SAMD12, which encodes the Sterile Alpha Motif Domain Containing 12 protein. Western blot analysis in available brain tissue showed slightly but significantly decreased SAMD12 expression levels when compared to controls, suggesting a possible role of the protein in disease. However, the authors suggest that the disease mechanism may lie at the RNA- rather than the protein-level, particularly since the two SAMD12-negative families were also found to have TTTCA- and TTTTA-repeat expansions in other genes. Examination of autopsied brain tissue identified RNA foci, containing UUUCA sequences but not UUUUA sequences, in cortical neurons and Purkinje cells in patients but not in controls. RNA foci result when an RNA molecule contains an expanded number of repeats. RNA molecules containing an extra number of repeats are retained in the cell’s nucleus, where the RNA molecules take on an abnormal structure and isolate other RNA binding proteins along with them. These aggregates – called RNA foci – have toxic effects within the cell. RNA foci have also been observed in other neurodegenerative disorders caused by repeat expansions, such as C9ORF72 in ALS/Frontotemporal Dementia and DMPK in myotonic dystrophy. These findings suggest that BFAME is a so-called “RNAopathy”, and that RNA-mediated toxicity of RNA products containing UUUCA sequences may be the underlying pathomechanism.
Implications for other epilepsies. The findings from this study are fascinating and immediately lead us to question what we can learn from them. The non-coding pentanucleotide repeat expansions identified in patients in this study would likely not be detected by short read sequencing or whole exome sequencing, which has become the standard diagnostic and research-based sequencing methodology currently. Could intronic repeat expansions be playing a role in other unsolved presumed genetic epilepsies, including the genetic generalized epilepsies? Although we are increasingly successful in identifying underlying genetic causes in patients with severe, early-onset epilepsies, we may need to explore new methodologies – including whole genome sequencing and/or RNAseq – to crack the difficult to solve cases of the future.
What you need to know. BFAME1, linked to 8q24, is due to intronic TTTCA- and TTTTA-repeat expansions within the SAMD12 gene. Larger repeat sizes are correlated with earlier age of onset. Expression of RNA containing these repeat expansions likely lead to RNA foci in cortical neurons and Purkinje cells, suggesting that BFAME is an RNAopathy and that the underlying pathomechanism is related to RNA-mediated toxicity of RNA products containing the repeated sequences.