
800 years. The discovery of SCN1B as a causative gene for Genetic Epilepsy with Febrile Seizures Plus (GEFS+) was one of the most pivotal moments in epilepsy genetics. This discovery not only shaped our understanding of the channelopathy concept, but also highlighted the importance of careful phenotyping. Therefore, it may be surprising that SCN1B took almost a quarter of a century to accrue sufficient evidence to be considered as a definite epilepsy gene. However, this is not the only aspect where SCN1B operates on its own time scale. In a recent publication, one of the most common disease variants in SCN1B could be traced back more than 800 years to a single founder event. Here is a 2023 update on the journey of one of the most well-known but also most mysterious epilepsy genes whose origins are lost in the depth of time.
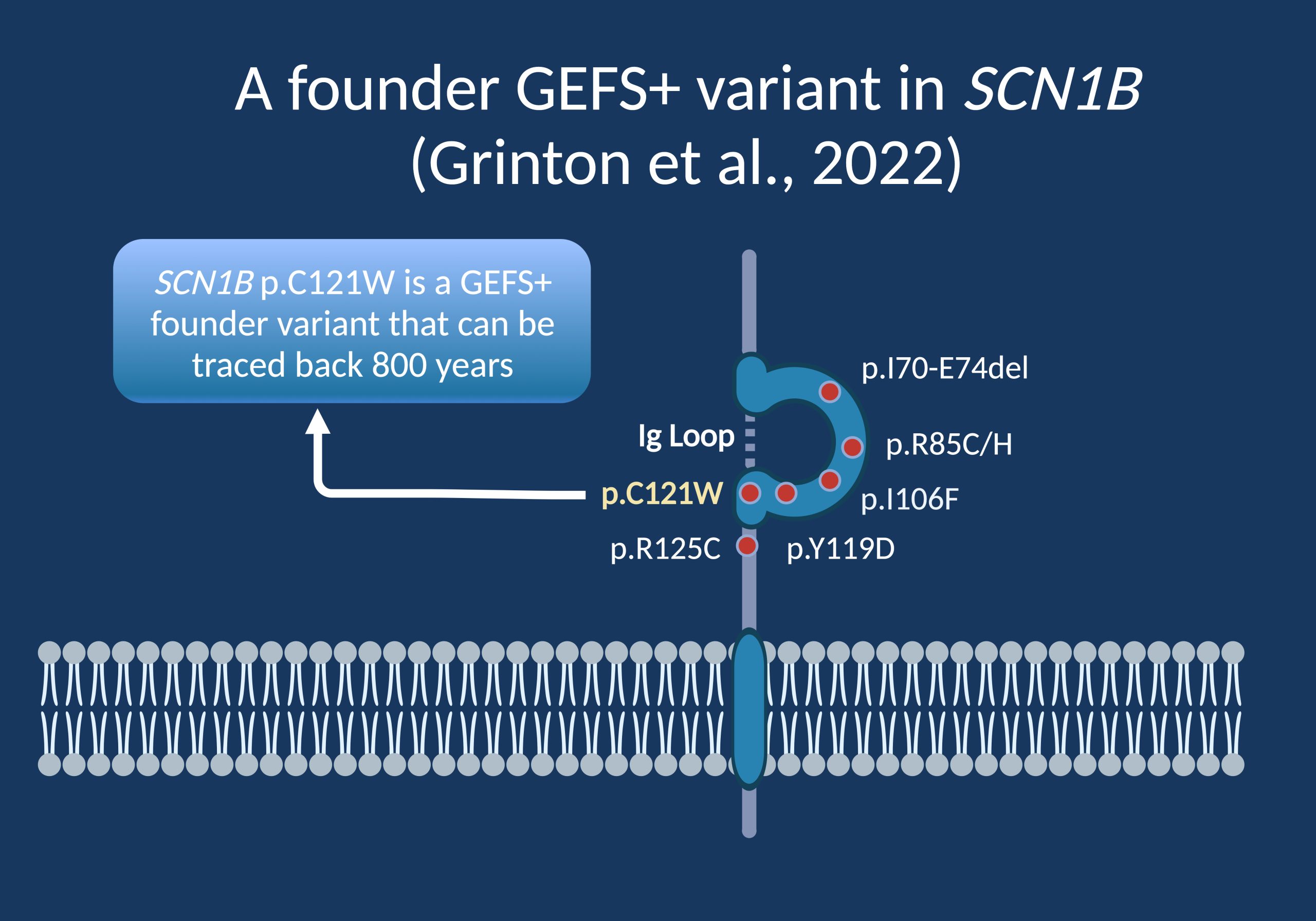
Figure 1. Despite its designation as a sodium channel subunit, the SCN1B protein is not a channel protein itself. While the alpha subunits of voltage-gated sodium channels have six transmembrane segments, the auxiliary beta subunits modulate the biophysical properties of sodium channels. All beta subunits are composed of an N-terminal immunoglobulin-like (Ig) domain and a transmembrane region. The vast majority of disease-causing variants in SCN1B are located in the Ig Loop. [created with BioRender]
Heterogeneity. Distinguishing between simple febrile seizures (FS) and FS+ was crucial in characterizing the true genetic burden in GEFS+ families. In fact, our 2008 review that gave the name to this blog used the original SCN1B GEFS+ pedigree as an example on why phenotyping is so critical. Within the large multiplex pedigree with the causative SCN1B variant, there are entire branches of the family with FS that do not carry the causative variant, highlighting that FS and FS+ are conceptually different while sharing common clinical features. Without developing the GEFS+ concept, the causative gene in this family may have been elusive at the time.
Gene validity. The serendipity of gene discovery despite the phenotypic heterogeneity, phenocopies, and incomplete penetrance of GEFS+ pedigrees provided a challenge on a different level. Putting on my hat as the co-chair of the ClinGen Epilepsy Gene Curation Expert Panel (GCEP), the SCN1B gene simply could not accrue sufficient evidence to have it become a definite epilepsy gene. This may sound strange, but assessing gene validity (i.e., if a gene is a true disease gene) is a standardized, agnostic process that meticulously picks apart the available evidence for each gene. This process is crucial – if we would like to move from gene discovery to precision medicine, we need to have strict and agreed-upon criteria on what makes a gene a true disease gene. Until December 2022, SCN1B simply did not pass the threshold yet.
Recessive. Let me restate the prior sentence: the autosomal dominant GEFS+ phenotype did not pass the threshold. SCN1B could easily be confirmed to be a definite genetic etiology for the recessive developmental and epileptic encephalopathy (DEE) phenotype. Accordingly, for the majority of our GCEP’s existence, SCN1B was in limbo. The recessive presentation was definite, while the autosomal dominant presentation simply did not yield enough evidence to pass the threshold as a definite genetic etiology. Gene validity is based on peer-reviewed publications and finally, in December 2022, we were able to assign “definite” status to the autosomal dominant GEFS+ presentation. SCN1B is a unique example on how phenotypic heterogeneity may interfere with assessing gene evidence – it took SCN1B almost a quarter of a century to become a definite epilepsy gene.
The millennium variant. SCN1B is aeonic in other aspects, as well. In a recent publication in the American Journal of Human Genetics, Grinton and collaborators identified the SCN1B c.363C>G (p.C121W) variant, one of the most common disease-causing SCN1B variants as a founder variant that survived through weak selective pressure for 800 years. The authors examined 14 GEFS+ families carrying this “millennium variant” and identified the same haplotype in all families. In other words, the genetic backbone of this variant was identical in all families, suggesting a single founder event with the most recent ancestor living approximately 800 years ago. This means that the clinical presentation currently considered GEFS+ was already around at the time of Genghis Khan, the Inquisition, and Thomas Aquinas.
Back to 2022. Given the existing resources available to date, the authors tried to examine whether they can validate the existence of a single founder event in large biorepositories. When examining the UK Biobank, they identified an additional 74 individuals with the SCN1B c.363C>G (p.C121W) variant – again, all individuals carried the same haplotype, the same genomic signature suggestive of a single founder event.
Odds ratio. Interestingly, the availability of clinical information in the UK Biobank posed a unique challenge. Only five out of 74 individuals had a history of seizures, a remarkably low frequency for a dominant epilepsy gene, even if we account for incomplete penetrance and phenotypic heterogeneity. While the frequency of mild childhood epilepsies in the UK Biobank is likely vastly underreported, this raises an interesting question: where is the threshold between a monogenic condition and a risk gene? The authors calculated odds ratios for the SCN1B c.363C>G (p.C121W) variant despite the likely missing data in the UK Biobank. With estimates of an OR between 2 and 13, this number is reminiscent of the risk contributed by rare microdeletions such as 15q13.3 or 16p13.11.
The GEFS+ challenge. However, when reviewing the existing SCN1B GEFS+ pedigrees, SCN1B looks much more like a genetic etiology inherited in an autosomal dominant fashion rather than the counterintuitive pattern seen with epilepsy-related microdeletions. In summary, while the heterogeneity and incomplete penetrance allowed the SCN1B c.363C>G (p.C121W) variant to survive through dozens of generations, GEFS+ again challenges our understanding of autosomal dominant epilepsy syndromes as it initially did when the GEFS+ concept was initially conceived. GEFS+ and the phenotypic spectrum associated with SCN1B continues to be a fascinating topic, highlighting the aspects that make the field of epilepsy genetics so unique. In summary, the publication by Grinton and collaborators reinvigorated the interest in SCN1B-related GEFS+ and emphasized that epilepsy genetics in 2023 includes much more than understanding de novo variants or polygenic risk scores.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
