
Heterogeneity. The diversity of clinical presentations and responses to anti-epileptic drugs (AEDs) has posed a major obstacle in developing strategies to treat patients with SCN2A-related epilepsies. While the literature provides multiple examples of single case reports with favorable responses to various AEDs, the broad range of disease presentations and known or presumed effects on channel function has made it extremely difficult to extrapolate findings from one patient to another. In a recent publication in Brain, we reviewed the largest cohort of patients with SCN2A-related neurodevelopmental disorders so far, including a subset of patients with detailed phenotypic data over time. With this data, we were able to find support for the hypothesis that age of seizure onset correlates with the functional effect of the mutations and the response to common anti-epileptic medications, taking a first step towards understanding the SCN2A mystery.
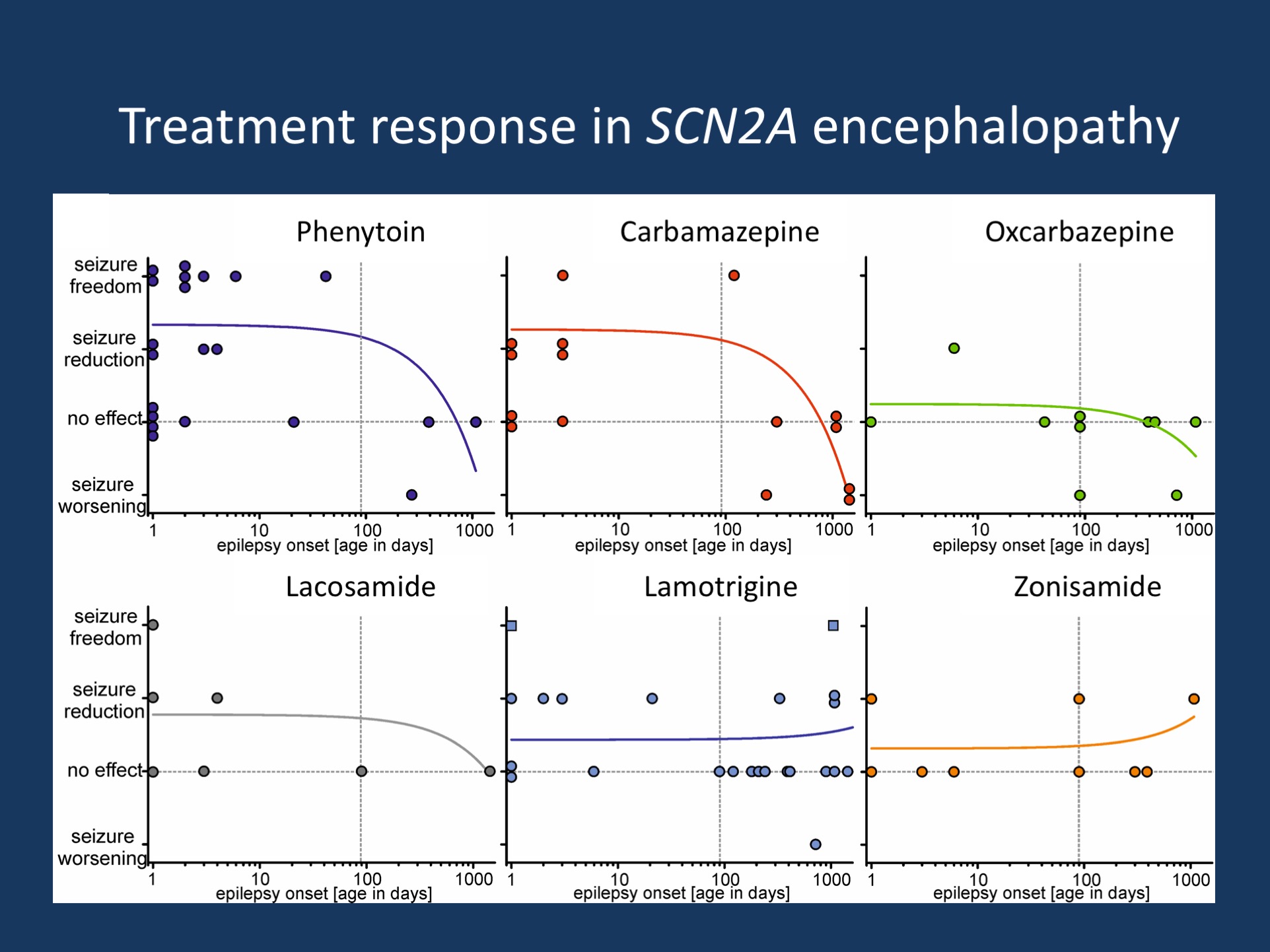
Figure. Treatment effects of common anti-epileptic medications in SCN2A-related disorders. Review of the treatment response in patients taking phenytoin, carbamazepine, oxcarbazepine, lacosamide, lamotrigine, or zonisamide suggests that the response to these anti-epileptic medications is related to age of seizure onset. These six AEDs are typically considered sodium channel blockers (SCBs) or have sodium-blocking properties. In a separate set of experiments, we were able to demonstrate that both the age of onset and the response to SCBs correlates with gain-of-function in early-onset cases and loss-of-function in patients with a later onset. (Figure reproduced from a non-copyrighted preliminary version of Figure 2B by Wolff and collaborators)
Disclaimer. In this blog post, I will only be able to point out some aspects of the publication by Wolff and collaborators. This publication is our big SCN2A publication that reviews the phenotypes of 200 patients with SCN2A-related neurodevelopmental disorders. In fact, the publication is dense and packed with sufficient data for several potential posts that focus on recurrent mutations, disease phenotypes, and functional data. In this post, however, I would like to discuss the main message of our publication: the correlation between age of seizure onset, mutational mechanism, and response to sodium channel blockers. To fully understand the complexity of this question, let’s go back to first discovery of SCN2A as a gene for human epilepsy.
From BFNIS to EE. The discovery of SCN2A in human neurodevelopmental disorders occurred somewhat in reverse order. The familial syndrome of Benign Familial Neonatal/Infantile Seizures was the first SCN2A phenotype to be described. Mutations were identified in large families where affected individuals had neonatal and/or infantile seizures and a normal neurodevelopmental outcome. This familial form of SCN2A has been reported in independent studies after the initial discovery, but it became obvious quickly that this familial syndrome is much less common than familial neonatal seizures due to mutations in KCNQ2 or infantile seizures due to PRRT2. With the publication by Wolff and collaborators, we are now able to put a number on this: only 15% of patients with SCN2A-related neurodevelopmental disorder have the benign phenotype, while four times as many patients have an epileptic encephalopathy phenotype.
SCN2A in autism. However, before the epileptic encephalopathy phenotypes were fully appreciated, SCN2A made an unexpected comeback in some of the first larger autism studies that used exome sequencing. SCN2A had suddenly become an autism gene, where loss-of-function mutations were observed. It was during this time that also the first reports of SCN2A in epileptic encephalopathies were appearing in the literature, even though it took several years for this picture to fully emerge. SCN2A was one of the first genes we reviewed on this blog, and back in April 2012 I was confused by the heterogeneity of this gene, sometimes even questioning whether SCN2A may be a disease gene at all.
Disease over time. The publication by Wolff and collaborators now adds an important piece to the puzzle. Basically, trying to paint the picture of SCN2A as a “autism gene” is somewhat incomplete. Patients with SCN2A truncating or loss-of-function mutations usually have epilepsy later in childhood, a phenotype that may have been missed in earlier autism-based exome studies. In fact, only 16% of patients with SCN2A-related neurodevelopmental disorders do not have seizures, suggesting that epilepsy is the most prominent feature of this condition.
Response to sodium-channel blockers. Understanding that the temporal aspect is important when looking at SCN2A-related epilepsies, we assessed the drug response in patients with respect to age of onset of the epilepsy. There have been previous reports that sodium channel blockers may be particularly useful in patients with SCN2A-related epilepsy, but we could show that this mainly applies to patients with an early age of seizure onset. In patients with a later age of seizure onset, these medications may even worsen seizures. Figure 2B in the publication by Wolff and collaborators that is reproduced in this blog post tells a somewhat clearer story. Even though the numbers are small, there is a clear trend for sodium channel blockers to improve seizures in early-onset epilepsies and have no effect or even worsen seizures in patients with a later onset. Grouping patients with different epilepsy syndromes and grouping different AEDs based on their joint function as sodium-channel blockers is somewhat hypothesis-driven and imprecise. However, it provides an interesting approach to overcome the vast genetic heterogeneity in some neurodevelopmental disorders and gives a useful template on how we can approach the genetic heterogeneity in other conditions.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.

Excellent as usual Ingo in clarifying things as you find them. I wondered if the drug that is the most useful for the type of seizure can be started at any age? Hope that makes sense. Thanks for your hard work
Dear Tracy,
thank you for your email. That’s an interesting question and not that was easy to answer! We would assume that this would probably be the case, i.e. that response to a certain medication is constant over time, but it is hard to draw this conclusion from the data given that longitudinal data is always difficult to obtain…