
Epilepsy gene panel. Testing for genetic causes in human epilepsy is typically performed using gene panels. In contrast to our research-based exome studies in an academic setting, much of the gene panel testing is performed through commercial laboratories and much of the existing data is usually inaccessible to the scientific community. In a recent publication in Epilepsia, a large US-based diagnostic laboratory reports on some of their existing data on epilepsy gene panels by reporting the results of more than 8500 epilepsy gene panels – a cohort size that is more than five times larger than any prior exome or gene panel study in the epilepsy field. I was asked to write an editorial on this publication, and I also wanted summarize on our blog three key messages that you can take away from this study.
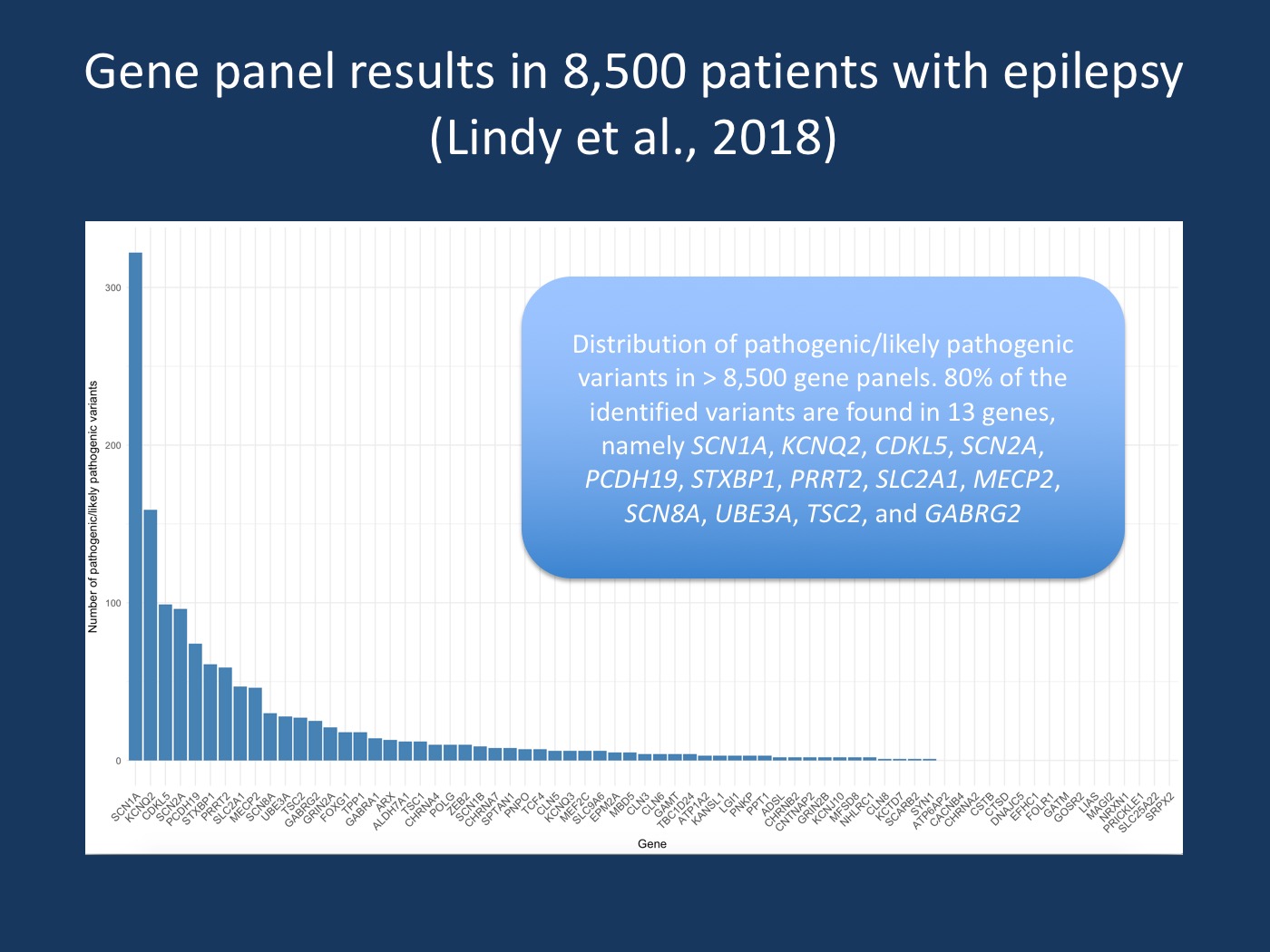
Distribution of genes. Distribution of pathogenic/likely pathogenic variants in > 8,500 gene panels. Four genes (SCN1A, KCNQ2, CDKL5, SCN2A) account for 50% of all pathogenic/likely pathogenic variants and SCN1A alone accounts for almost one fourth of all variants. 80% of the identified variant belong to only 13 gene, including SCN1A, KCNQ2, CDKL5, SCN2A, PCDH19, STXBP1, PRRT2, SLC2A1, MECP2, SCN8A, UBE3A, TSC2, and GABRG2.
Summary.The study by Lindy and collaborators looks at the diagnostic results in 8565 patients with epilepsy and neurodevelopmental disorders who were tested via a gene panel of 70 genes for epilepsies and related neurodevelopmental disorders. The authors find pathogenic or likely pathogenic variants in roughly 15% patients and identify SCN1A and KCNQ2 as the most common genes. Of the 70 genes tested, only 22 genes had a high yield of positive findings, while 16 genes did not show a single positive finding. Here are three key messages of this publication.
1 – The diagnostic yield of panel testing is 15%
With a rate of pathogenic/likely pathogenic variants of 15%, the study by Lindy and collaborators lands within the lower range of what has been previously reported. While the authors comment on the fact that their cohort was unselected, the size of the cohort speaks for itself and represents somewhat of a landmark that puts other studies into perspective. 15% is a solid estimate for gene panel studies and everything above this estimate is either due to selected cohorts or the use of other testing options.
2 – Major genes
The distribution of pathogenic and likely pathogenic variants across the 70 genes tested in the study by Lindy and collaborators looks like a Pareto distribution (80/20). More than 80% of all variant are explained by the top 20% genes. SCN1A alone almost accounts for one fourth of all pathogenic/likely pathogenic variants; the top four genes (SCN1A, KCNQ2, CDKL5, SCN2A) account for a little more than 50% of all variants. A small 25-gene panel would have captured more than 90% of all variants, accounting for a diagnostic yield of 14% in the larger cohort. This observation argues for the use of smaller gene panels as a first-tier screening given that a good amount of the genetic diagnoses that will be found by a panel within a limited number of genes.
3 – Genes without evidence
While there are genes with a very high number of variants, the study by Lindy and collaborators also identified a large number of genes without any pathogenic or likely pathogenic variant. These genes include ATP6AP2, CACNB4, CHRNA2, CSTB, CTSD, DNAJC5, EFHC1, FOLR1, GATM, GOSR2, LIAS, MAGI2, NRXN1, PRICKLE1, SLC25A22, and SRPX2. In my commentary, I provided two explanations for this observation. In fact, some of these genes may be extremely rare and may be tested in patients with a clinical diagnosis through other means. This applies to the established genes for Progressive Myoclonus Epilepsies including CSTB and GOSR2. It is also important to mention that the NGS methodology used in this study does not detect the most common pathogenic variant in CSTB, a dodecamer repeat expansion. On the other hand, some of the genes without identified pathogenic/likely pathogenic may not be disease genes at all. These genes were once thought to be good candidates, but further evidence for pathogenicity has not emerged. This applies to genes such as EFHC1, MAGI2, or SRPX2. These genes will probably be removed from diagnostic panels in the near future given the lack of evidence for a disease association. Basically, these genes are not disease genes and holding on to them in an indefinite candidate status despite lack of confirmatory evidence will eventually create more harm than good.
What you need to know. With the current study by Lindy and collaborators, the epilepsy field has taken a leap forward. Testing 70 genes in more than 8,500 patients with epilepsy provided clear evidence for common epilepsy genes versus unconfirmed candidates and indicated that up to 15% of patients can receive a genetic diagnosis based on testing of a limited number of genes. There are other aspects of this study that I did not include in this post and may discuss in the future, namely the role of deletion/duplication testing for each gene, recurrent variants, and the rate of inherited versus de novo variants.
Caveats. One important issue to mention is that some genes that are presumably not all that rare were not included in the study by Lindy and collaborators. For example, CHD2 and DEPDC5 were not among the 70 genes analyzed in their study. A further limitation of the publication by Lindy and collaborators is the lack of detailed phenotypic data. However, the size of their study alone provided important insights into the genetic architecture of human epilepsy.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
