
Dravet Syndrome. In 2011, our EuroEPINOMICS-RES program was in full swing. We had recruited a cohort of 31 patients with Dravet Syndrome who had been previously tested negative for mutations in SCN1A with the aim to identify novel genes for this epileptic encephalopathy. Even though this cohort was crucial in our identification of CHD2, HCN1, and KCNA2 as novel genes for genetic epilepsies, the main finding in this cohort was something that we did not expect. Roughly one third of our 31 patients had mutations in SCN1A, even though they had previously been tested negative. In a recent publication in Molecular Genetics and Genomic Medicine, we tried to understand what had happened and joined forces with other groups who had made the same observation. Here is the story of the missed SCN1A mutations.
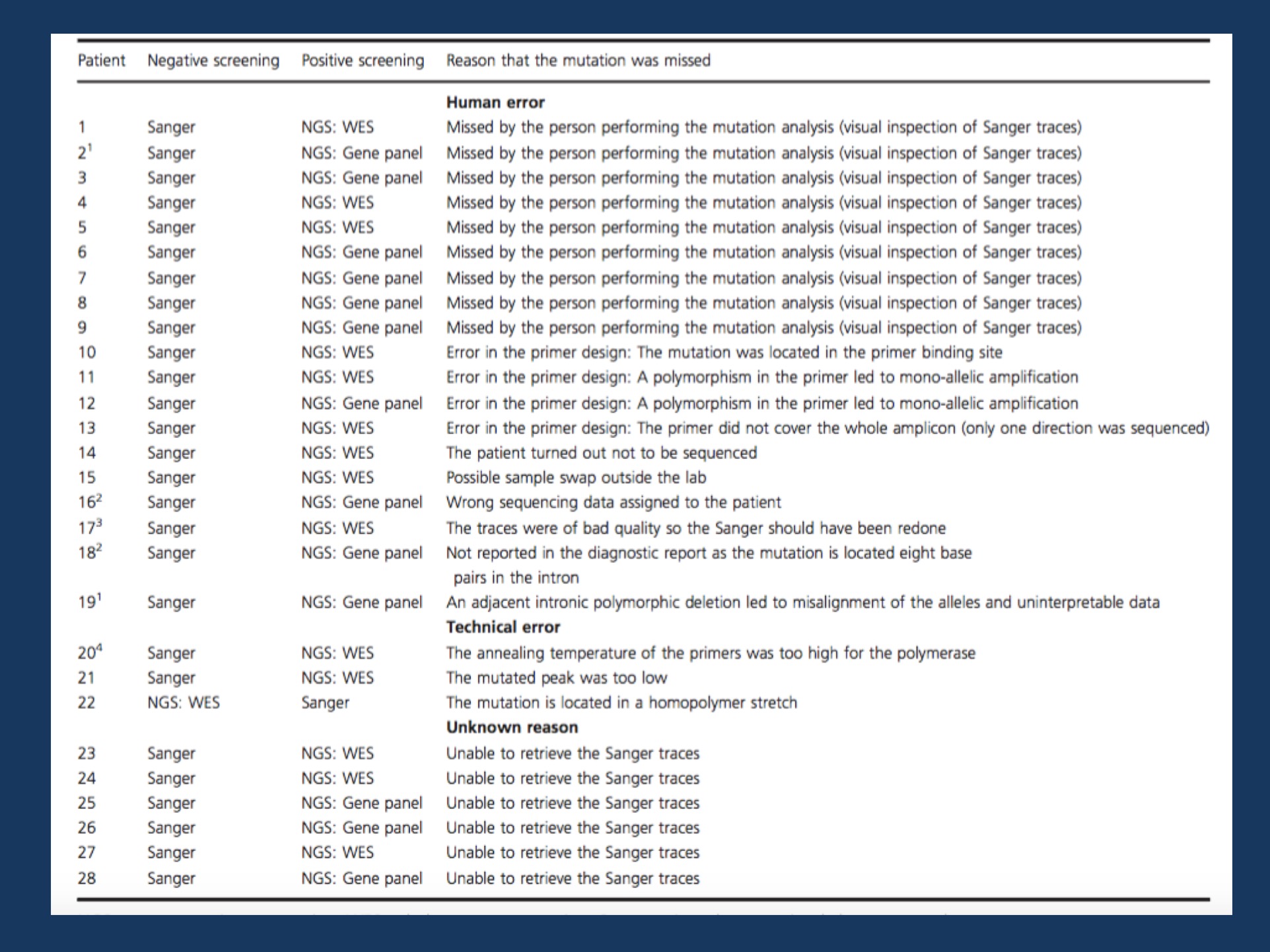
History of 28 SCN1A mutations that were initially missed, but then discovered through further genetic testing on a clinical or research basis. Image adapted from the publication by Djemie and collaborators under a Creative Commons Attribution License (CC BY).
Background. First of all, let me correct the title of this blog post. This post and our recent publication is the story of missed SCN1A mutation in Dravet Syndrome. It is a story on how phenotype trumps genotype in modern genomic medicine, a story that is ultimately reassuring to me as a clinician who emphasizes that genetics only helps us in understanding epilepsies, but does not define it. It reinforces the notion that Dravet Syndrome is a clinical diagnosis, not a diagnosis you get with a genetic test.
Phenotype. Traditionally, the severe epilepsies of childhood have been very difficult to characterize, as many patients have unique and sometimes unusual features. Within this heterogeneous group of disorders, Dravet Syndrome has emerged as one of the best defined and most extensively studied entities. Dravet Syndrome is characterized by the onset of fever-associated seizures in the first year of life, which are often prolonged. As the disease progresses, afebrile seizures are seen including myoclonic seizures, atypical absences, and focal seizures. Seizures usually are resistant to currently available treatments, and the development of new therapeutic options for patients with Dravet Syndrome is an active field of clinical research. The psychomotor development of patients with Dravet Syndrome is initially normal. During the second year of life however, developmental delays frequently become apparent. In contrast to many other genetic epilepsies, there is a very strong connection between the clinical picture of Dravet Syndrome and the presence of a pathogenic variant in the SCN1A gene. More than 80% of patients with Dravet Syndrome have a pathogenic change in the SCN1A gene.
Missed mutations. In our recently publication by Djemie and collaborators, we were able to evaluate 28 SCN1A mutations that were initially missed, but then identified through other technologies. In most cases, patients had initially undergone Sanger sequencing, the classical sequencing technology for single genes. Most missed mutations were then discovered through either gene panel or exome sequencing. Why were these mutations missed in the first place? The most frequent reason for reporting false-negative results was human errors, which was the case in 66% of missed SCN1A mutations. Mutations were overlooked in the initial Sanger sequencing traces in 9 cases, but there were also other reasons why these mutations were missed including faulty primer design, sample swaps, and incorrect documentation. In three cases, technical errors could be attributed including one case where a mutation was initially missed through exome sequencing and later on discovered through Sanger sequencing – the only case in our series where a next generation sequencing technology missed a mutation that traditional Sanger sequencing picked up.
What does this story tell us? I think that there are three main lessons that the story of the missed SCN1A mutations tell us. First, much has happened since the first SCN1A sequencing effort included in our study and the current level of genetic diagnostics. More diagnostic laboratories operate by a high standard that aim at minimizing human error, which was the main contributor to the fact that SCN1A mutations were missed in the first place. Secondly, and more importantly, the story of missed SCN1A mutations shows us the limitations of Sanger sequencing. Sanger sequencing can be considered an analogue technology as both human visual inspection and automated detection needs to detect the height of a sequencing peak. In previous blog posts, we have given examples on how Sanger sequencing can be limited in some cases in patients with SCN1A variants and different genes. Next generation sequencing, in contrast, is a digital technology, where the assessment of signal intensity is measured in distinct units (reads). Conceptually, a digital technology is much less prone to errors that an analogue technology. Third, and most importantly, the story of the missed SCN1A mutation tells us about the genetic architecture of Dravet Syndrome. Even though our data may be incomplete and not representative of the current diagnostic technology, a patient with Dravet Syndrome has a reasonable chance of being positive for a mutation in SCN1A even though s/he was initially tested negative.
The clinical diagnosis. The connection between phenotype and genotype is so strong that even SCN1A-negative cases are much more likely to be due to SCN1A than to any other gene. Again, this observation reinforces that Dravet Syndrome is a recognizable clinical diagnosis. The clinicians contributing the patients to our study were able to make a diagnosis of Dravet Syndrome and –more importantly– were able to maintain the clinical diagnosis in the absence of initial confirmatory testing. This is strong proof of how aware clinicians around the globe have become of the clinical features of Dravet Syndrome.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.

This! I’ve seen the same situation where we identified a causal mutation in WES while reported negative. When we went back to old sanger traces, they were there as clear as they could be. Not sure if labs have multiple checkpoints where two or three techs take a look at the same data.
“Conceptually, a digital technology is much less prone to errors that an analogue technology”… Well conceptually. I kinda agree even though it brings other kinds of problems to the table.
Good report!
Thanks! I think SCN1A and Dravet is just a very drastic example as the connection between gene and phenotype is so strong. You’re right that NGS technologies may have other aspects to consider that we may not be fully aware of yet.
Hi Ingo,
This is really interesting to read. We (Sanger) are currently reassessing the transcript structure of msny EIEE genes, including SCN1A. We have found numerous novel exons that are not represented in exome panels and are in the process of trawling patient data to see if these new exons find novel mutations. It appears that current gene models in Ensembl and UCSC browsers are still missing crucial exons, which might be another reason that patients who present with a specific disease phenotype test negative for mutations.
Dear Charles, thank you for your comment. I agree that this is one of the mechanisms why patients can be mutation-negative. In our series, for example, we had one patient with an intronic mutation that was a bit further into the intron than most labs at the time had looked at. Therefore, in SCN1A and in many other genes, additional coding regions and non-coding regions are interesting targets to look at. This is an issue that will definitely come up when we advance from exome to genome sequencing.