
SCN1A. Finally, after various other epilepsy genes have been added, we are trying to put together a static website on SCN1A rather than updates only. SCN1A is by far the most prominent epilepsy gene and the first genetic etiology that comes to mind for anything relating to fever and seizures. While our Epilepsiome page will give you all the relevant facts regarding this gene, here is my personal view on the SCN1A story.
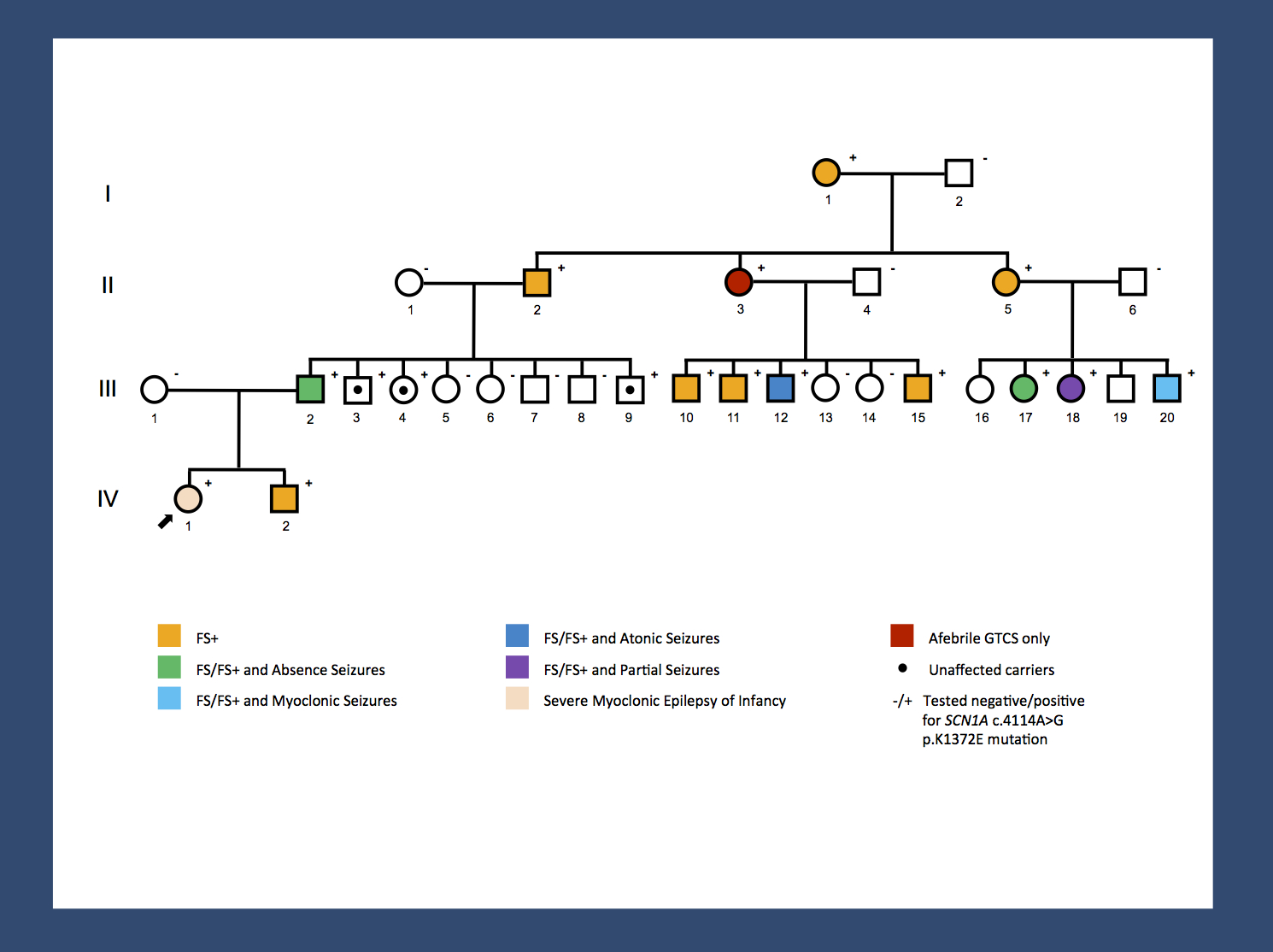
Re-posting one of our figures from November 2013 – a pedigree of a large GEFS+ family with 14 affected and 3 unaffected carriers published by Goldberg-Stern and colleagues. A missense mutation results in a broad range of phenotypes spanning unaffected carriers, Febrile Seizures Plus (FS+) phenotypes and Dravet Syndrome. The range of phenotypes seen in this family suggests that SCN1A is not the entire story. It will be interesting to investigate whether the modifying factors are genetic or non-genetic. The pattern of phenotypes suggests some intra-familial factors. (Disclaimer: I am a coauthor on the publication by Goldberg-Stern and colleagues).
The gene that changed …everything. By the time that I started working in epilepsy genetics, SCN1A had long been established as a prominent gene for human epilepsies. It all made sense back then – SCN1A was an ion channel gene both involved both in severe human epilepsies such as Dravet Syndrome and in milder familial epilepsies. SCN1A turned out to be the most convincing proof of the channelopathy concept, the idea that human epilepsies are intrinsically linked to ion channels. Genetics and functional studies finally seemed to converge. However, the SCN1A story was much more complex than this. In fact, there were three twists in the SCN1A story that tell us a bit about how the balance of genetic functional studies interact – and what happens to this balance in the setting of exponentially increasing genetic data. Looking back, this is how our thinking about SCN1A changed throughout the last 10 years.
Twist #1 –gain or loss of function. The question of whether SCN1A mutations were gain-of-function or loss-of-function was not solved through additional functional studies, but by increasing genetic data including the identification of truncating mutations and deletions. With functional data inconclusive, it was the recognition of partial SCN1A deletions that eventually made the story even clearer. There are publications as late as 2004 that claim that SCN1A mutations in Dravet Syndrome may be at least in some cases activating. After that, you hear very little about a potentially activating effect of some SCN1A mutations, suggesting that the increasing genetic data has made the case for haploinsufficiency much clearer than before.
Twist #2 – finding a voice for Dravet Syndrome. The SCN1A story is not only a story about an ion channel involved in epilepsy – it is also a story about re-defining a disease and the power of having a name for a disease. I deeply admire the work of the Dravet Syndrome Foundation and how they helped raise awareness for Dravet Syndrome. In 2015, every first year resident rotating through neurology knows about Dravet Syndrome – knowledge about this genetic epilepsy has become part of the basic neurology and pediatric training. This is also the reason why Dravet Syndrome is also the first genetic epilepsy for novel antiepileptic medications such cannabidiol. This is a motivating example for what we can do for many of the other genetic epilepsies.
Twist #3 – the return of the ion channels. Here’s a confession. I didn’t really believe in ion channels when I started working in the field. I thought that their time was over. Hence the name of the blog (“Beyond the Ion Channel”) and the title of our 2008 Lancet Neurology review. Initially, it seemed that I was not that far off – there were few new disease-causing ion channels discovered in the decade after SCN1A was found. However, in 2015, nothing could be further from the truth – large data turns up ion channel mutation after ion channel mutation and KCNQ2, KCNT1, SCN2A, SCN8A are relatively common genes in genetic epilepsies. Other genes like KCNA2 or CACNA1A are less frequent, but also add to the ion channel hypothesis. Therefore, even the greatest skeptic of the channelopathy concept would be convinced by now. We are far away from being “Beyond the Ion Channel”.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
