
SCN1A phenotypes. Readers of Beyond the Ion Channel will know that we often post about SCN1A, one of the first discovered and most common genetic causes of epileptic encephalopathy. We more or less assume that we understand the phenotypes associated with pathogenic variants in SCN1A: most commonly Dravet syndrome, which is associated with de novo variants, and less commonly genetic epilepsy with febrile seizures plus (GEFS+), associated with inherited missense variants. However, a recent publication by Sadleir and colleagues suggests that the phenotypic spectrum of SCN1A-related disorders may be broader than we have previously appreciated. Are there SCN1A-related epileptic encephalopathies in addition to Dravet syndrome?
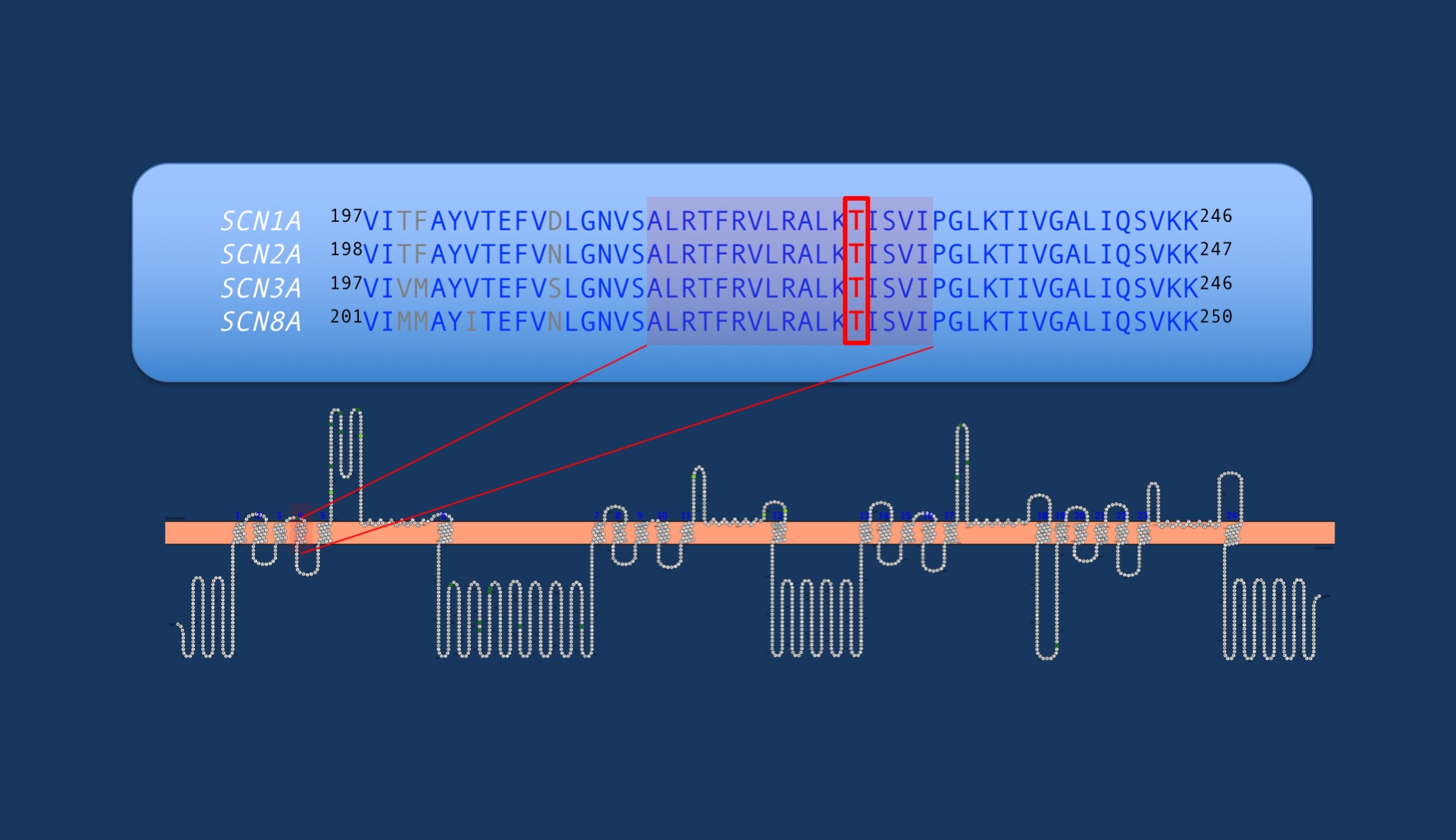
Figure. The p.T226 amino acid residue in SCN1A is part of a highly conserved region across voltage-gated sodium channels. This region forms the S4 transmembrane segment of domain I, which forms the voltage-sensor. So far paralogous variants in this Threonine amino acid residue have not been reported in other voltage-gated sodium channels, including SCN2A and SCN8A.
Beyond Dravet syndrome. Dravet syndrome is and remains a clinical diagnosis, and we are able to identify an underlying pathogenic SCN1A variant in >90% of patients. An SCN1A variant found in a patient who does not have classic features of Dravet syndrome would likely be treated with some degree of suspicion. However Sadleir and colleagues present compelling evidence that there may be a broader spectrum of SCN1A-related disorders beyond Dravet. In their study, they identify a recurrent SCN1A missense variant c. 677C>T p.(T226M) in 8 children with an early-onset epileptic encephalopathy much more severe than Dravet syndrome. These patients develop seizures before 12 weeks, often presenting with hemiclonic seizures or epileptic spasms, develop a severe hyperkinetic movement disorder, and have profound intellectual disability. Clinically, these children sound more similar to other genetic epileptic encephalopathies rather than classic Dravet syndrome.
Similarities and differences. There are some similarities to Dravet syndrome, though. Patients are described as having temperature sensitive seizures, hemiclonic seizures, and episodes of febrile status epilepticus. Important clinical differences are obvious, however. In addition to the earlier age of onset – Dravet syndrome typically presents after 4 months – patients with the recurrent T226M variant showed seizure types not typically associated with Dravet syndrome in childhood, including epileptic spasms and tonic seizures. Developmental outcomes in Dravet syndrome are generally milder than the patients described in this study, who were non-ambulatory and non-verbal. Hyperkinetic movement disorders, including chorea and dystonia, are also not associated with Dravet syndrome.
Genotype-phenotype correlations. Can we predict a patient’s phenotype based on the SCN1A variant? This is still very difficult to do. Truncating variants in SCN1A are typically associated with Dravet syndrome, but missense variants can be found in both Dravet syndrome and GEFS+. However the T226M variant appears to be associated with a non-Dravet severe epileptic encephalopathy. Sadleir and colleagues report another child with a similarly severe phenotype and a de novo P1345S variant. Another patient has been previously reported with a de novo V422L variant and phenotype that may be consistent with the severe SCN1A-epileptic encephalopathy, suggesting that the phenotype may not be restricted to the recurrent T226M variant.
Distinct disease mechanism? Although over 1200 variants in SCN1A have been reported, fewer than 20% are recurrent. The presence of the recurrent T226M missense variant in so many patients with a similarly severe phenotype is striking. Dravet syndrome is known to be associated with SCN1A haploinsufficiency. The presence of a recurrent missense variant suggests that perhaps this severe epileptic encephalopathy may be associated with a different disease mechanism, perhaps gain-of-function, as in SCN2A– and SCN8A–related disorders, or possibly a dominant-negative mechanism. Further functional studies are needed to further delineate whether the more severe SCN1A-epileptic encephalopathy is related to a different effect on Nav1.1 channel function.