
PURA. The title of this blog, Beyond the Ion Channel, is intended to reflect the wide variety of genes that can cause epilepsy and related neurodevelopmental conditions. Our last post on CACNA1A brought us back to channelopathies, so this blog post will again shift our focus. This post will introduce the new gene page for PURA, a gene that we did not feature as prominently as we should have. Here are five things to know about PURA, which is relatively recent to be described as a condition, and is likely more common than originally thought.
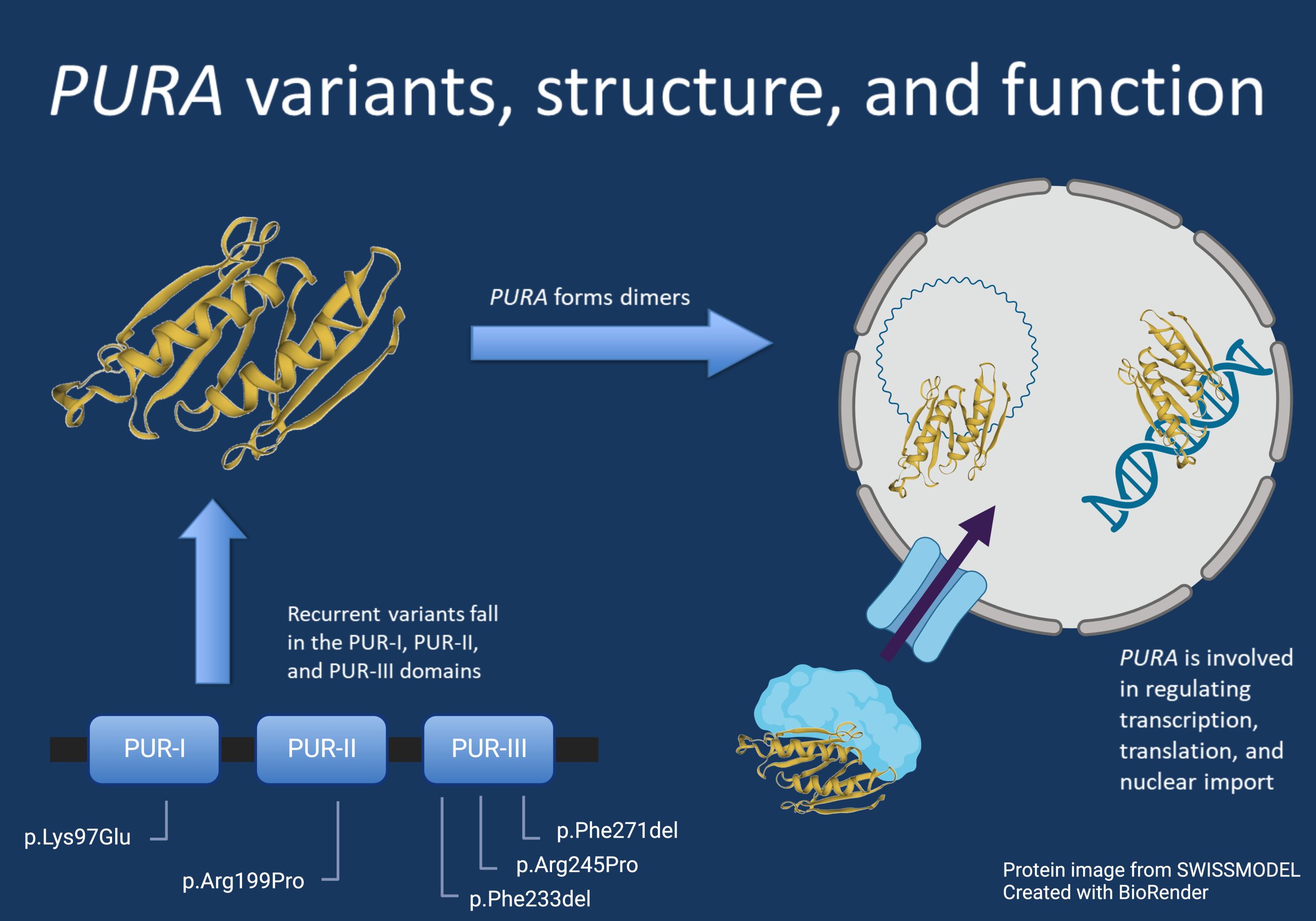
Figure 1. This figure shows the recurrent variants in PURA described in this page, as well as the overall structure of the protein and some examples of its functions in the cell. These functions include regulating genes and RNA and nuclear import.
1 – Breaking the pattern. For many genes associated with neurodevelopmental conditions, the initial reports of individuals tend to represent the more severe end of the spectrum associated with that condition, as these severely affected individuals tend to present for genetic testing first. As more individuals undergo genetic testing, this spectrum typically widens to include more mildly affected individuals. However, the wide disease spectrum associated with heterozygous, pathogenic variants in PURA has been evident since the first gene-disease association in 2014. Most individuals in the first cohort did not have seizures, and some had relatively mild developmental delay. As more individuals have been diagnosed with PURA over time, it has become apparent that epilepsy is in fact a more prominent feature. However, the disease spectrum remains relatively wide.
2 – Early disease course can be misleading. The early presentation of PURA-related neurodevelopmental disorders, particularly neonatal apnea and neonatal and possibly profound hypotonia, are features that can be suggestive of other common genetic neurological conditions such as spinal muscular atrophy (SMA), Prader-Willi syndrome, or Pitt-Hopkins syndrome. The early differentiation between these diagnoses is critical, as management and neonatal course is different for each of these conditions. This again highlights the importance of early, broad-based genetic testing to quickly end the diagnostic odyssey for individuals with neurogenetic conditions.
3 – Genotype-phenotype correlation. In many neurogenetic conditions in general, we expect some degree of genotype-phenotype correlation. However, this has not been observed in PURA-related conditions to date. This does not necessarily mean that there is no role of genotype in the associated phenotype of a given individual. Our previous work in HPO-based analysis of STXBP1 is a case in point. Through this work, particular genotype-phenotype correlations for recurrent variants were uncovered in a condition where this was previously thought not to be the case. In general, this standardized phenotyping approach in large cohorts may be an important step in fully elucidating genotype-phenotype correlation.
4 – Gene discovery pathway. The Deciphering Developmental Disorders study (DDD), which involved exome sequencing for over four thousand trios, greatly enriched our understanding of epilepsy genes that were known at the time as well as genes that had emerging relationships with human disease. PURA was one such gene. This study is still an essential resource we use today to inform our understanding of candidate genes.
5 – Animal models – what they can and cannot tell us. We have multiple tools to understand neurogenetic conditions in our toolbox, one of which is animal models. When we evaluate these in a formal setting, such as our ClinGen Epilepsy Gene Curation Expert Panel, we must consider two major factors – 1. Does the genetic mechanism required to elicit a phenotype in an animal model mirror what is seen in humans? and 2. Does the phenotype seen in a given animal model match what is seen in humans? For PURA, current mouse models meet the second criterion but not the first, as only mice with homozygous knockout show a phenotype. Because of this, when evaluating studies using these models few must consider the relevance to a human context.
Here is what you need to know. PURA-related disorder has emerged as a perplexing genetic epilepsy with a disease mechanism that is poorly understood thus far, particularly the molecular mechanism. The phenotypic spectrum is notable for several features that are quite prominent, such as the profound neonatal hypotonia. With better recognition and improved understanding of the natural history and disease course, we hope that this largely underdiagnosed condition may soon be a candidate for precision medicine approaches.