
Shaw. It has been a while since we have written about novel gene discoveries in the epilepsies, so I wanted to start this blog post with a general introduction to the genes that are still undiscovered, waiting to be identified. Currently, we assume that there are several hundred genetic etiologies for human epilepsies “out there” that we have not characterized yet. One of the most recent members to join the group of epilepsy genes is KCNC2 that we described in a recent publication. KCNC2, coding for a member of the Shaw-related voltage-gated potassium channels, presents with a phenotypic spectrum that is different from many other epilepsy genes.
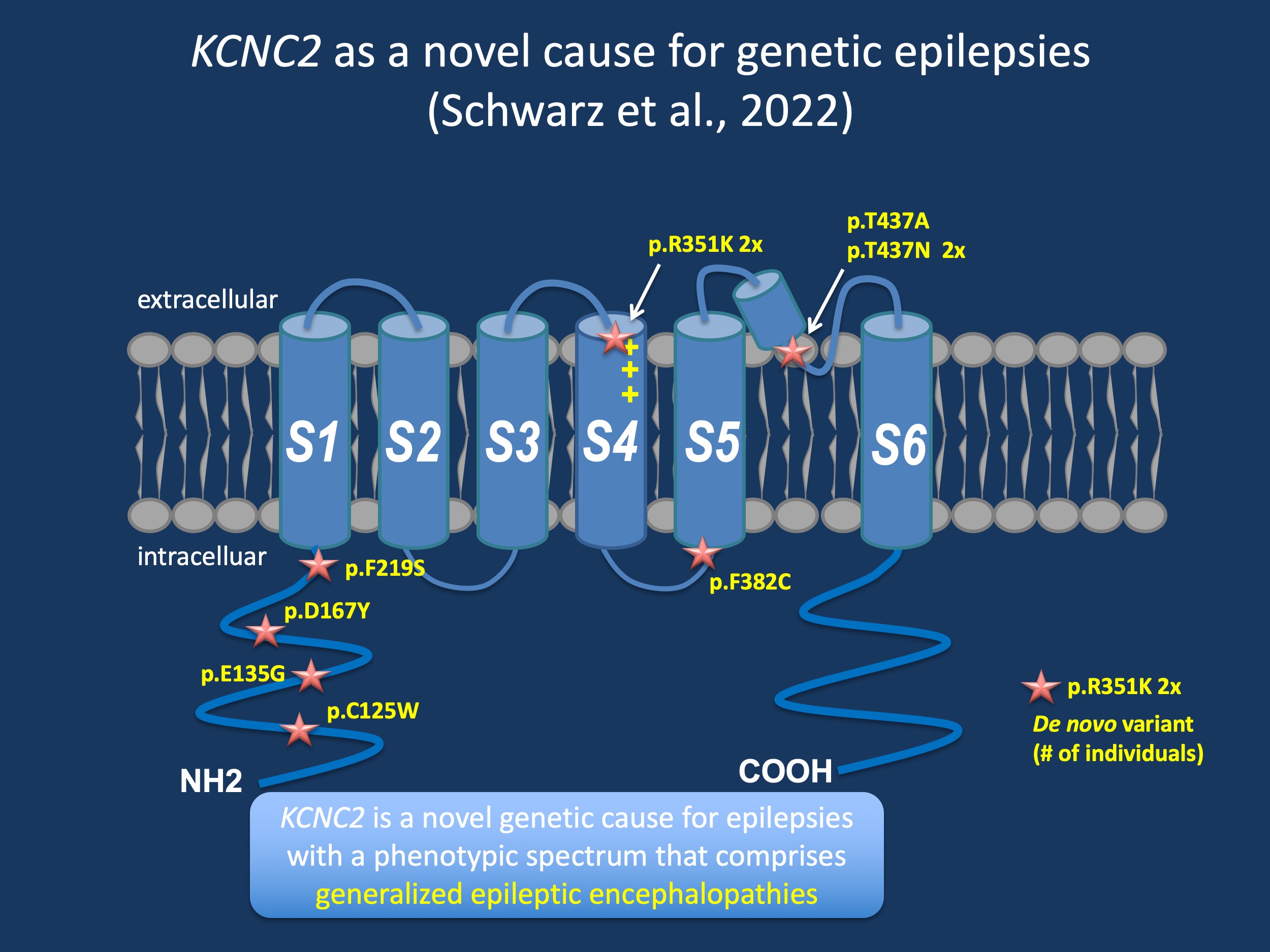
Figure 1. Structure of the KCNC2 channel and de novo variants identified in individuals with epilepsy. KCNC2 is new epilepsy gene with a phenotypic spectrum than includes early-onset generalized epilepsies. The KCNC2 channel belongs to the Shaw-family of potassium channels, which are involved in regulating neuronal action potential duration and firing patterns. The KCNC2 channel, also referred to as Kv3.2 is mainly expressed in CNS interneurons.
Potassium channels. Let me start with a brief overview of human potassium channels. In contrast to sodium or calcium channels there are quite a few of them, 79 human potassium channel genes, to be precise. Several members of this group have been implicated in human epilepsy, most prominently KCNQ2, KCNT1, and KCNA2. KCNQ2 is the causative gene for self-limiting familial neonatal seizures and KCNQ2-related developmental and epileptic encephalopathy (DEE), KCNT1 causes a DEE characterized by migrating focal seizures in infancy, and KCNA2 is a gene we initially identified through the EuroEPINOMICS consortium as a cause for DEE with ataxia. Despite the fact that all genes are ion channels permeable to potassium, their functions are quite different. KCNC2 belongs to the so-called “Shaw-family” of potassium channels, which are involved in regulating neuronal action potential duration and firing patterns. The KCNC2 channel, also referred to as Kv3.2 is mainly expressed in CNS interneurons.
KCNC1. A closely related potassium channel is the KCNC1 channel, which was initially discovered as a cause for a progressive myoclonus epilepsy (PME) with a single recurrent variant (p.Arg320His) that was found in most individuals. Since the initial discovery, the phenotypic spectrum of KCNC1 has widened to a broader range of DEEs. The specific subtype of PME caused by KCNC1 is a ULD-type PME. As outlined in our initial blog post on KCNC1, the progressive myoclonus epilepsies are a group of seizure disorders that initially present with therapy-resistant myoclonus that gets worse over time and can be accompanied by generalized tonic-clonic seizures and cognitive decline. The PMEs are usually thought of as a distinct group of epilepsies with a genetic basis and are usually divided into two larger groups. Unverricht-Lundborg disease (ULD) usually has preserved cognition until late into the disease, while disorders such as Lafora body disease show prominent neurological decline and dementia. Only a few years ago, the genetics of ULD-type PME were simple, as an expansion of a dodecamer repeat in the 5’-UTR of the Cystatin B (CSTB) gene was the only known genetic cause. However, with the discovery of SCARB2, PRICKLE1 and GOSR2, additional genes were found in ULD-type PMEs. The Lafora-type PMEs were traditionally thought of as more heterogeneous as several storage disorders including sialidosis, mitochondrial disorders or neuronal ceroid lipofuscinosis may present as progressive myoclonus epilepsies.
The KCNC2 phenotype. In the recent publication by Schwarz and collaborators, we report on 18 individuals with variants in KCNC2, including 10 individuals with de novo variants. When I saw the phenotypic spectrum, two individuals stood out. Both Individual 1 (p.C125W) and Individual 8 (p.T437A) had Early Onset Absence Epilepsy (EOAE). EOAE is a very rare generalized epilepsy characterized by absence seizures and generalized EEG findings before the age of 4 years. To find this unusual phenotype in 2/8 individuals with EOAE is surprising and hints at a phenotypic spectrum that is clearly distinct from other DEEs. Individual 7 (p.F382C) had Myoclonic Astatic Epilepsy (MAE) or Doose Syndrome. As we discussed in a prior post, there are only few established genetic etiologies for MAE and KCNC2 is one of them. Likewise, 2/8 individuals had continuous spike and wave during slow sleep (CSWS), another DEE-related finding that is often presumed to be genetic, but where a definite cause is often not found. In addition to the de novo variants, 5/8 variants where inheritance could not be established were found in individuals with Genetic Generalized Epilepsy (GGE). In summary, the overall phenotypic spectrum of KCNC2 is unusual – we found more generalized phenotypes than expected, hinting at a phenotypic spectrum that is more reminiscent of the generalized phenotypes of GABRG2, GABRA1, SLC6A1 or GLUT1 than other genetic DEEs. In addition, the spectrum is clearly distinct from the PME phenotype that is associated with KCNC1.
What you need to know. KCNC2 is a new epilepsy gene, a new epilepsy-related potassium channelopathy. With a phenotypic spectrum of de novo variants emerging that span the DEEs, there is an unusual preponderance of generalized DEEs including early-onset absence epilepsies and myoclonic-astatic epilepsy. This tendency to cause generalized epilepsies may reflect the particular role of KCNC2 on neuronal firing in fast-spiking interneurons. Better understanding the genetic and phenotypic spectrum of KCNC2 may help delineate better personalized treatment strategies.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
