
EEs. The concept of epileptic encephalopathy refers to a process where epileptic activity impairs overall brain function, including cognitive function, language, and behavior. In a recent commentary in Epilepsia, our current use and misuse of the concept of epileptic encephalopathy is reviewed critically. In summary, the authors criticize that epileptic encephalopathy is used as a diagnostic category rather than a description of the actual epileptic process, suggesting that another term may be necessary for the group of patients with intellectual disability and epilepsy where we often find a genetic etiology. In this blog post, I would like to plead guilty on behalf of the epilepsy genetics community for having misused the concept of epileptic encephalopathies for almost a decade. And we have done this for at least three different reasons.
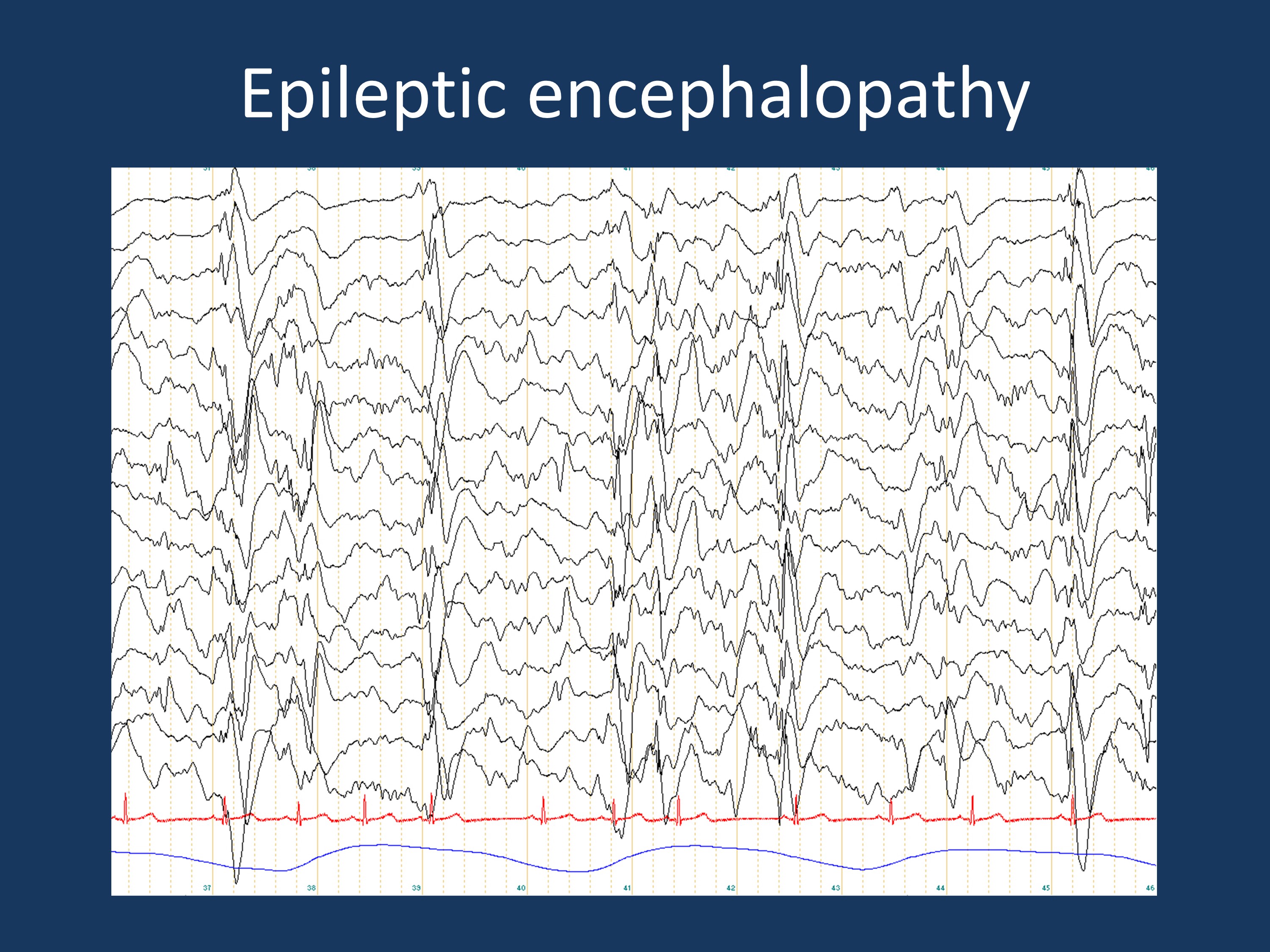
EEG findings in a patient with Lennox-Gastaut Syndrome. The EEG shows the finding of continuous slow spike-wave activity, indicating a profound disturbance of cortical function leading to an epileptic encephalopathy. The current blog post discusses whether the concept of epileptic encephalopathy should be used as a diagnostic category or should be reserved for a particular state of the patient’s disease.
Epileptic encephalopathy. In 1841, the general practitioner W.J. West from Tunbridge, England was the first person to report on a condition that eventually came to bear his name: West Syndrome. Dr. West reported the symptoms in his own son, including his attempts to treat the the “peculiar form of infantile convulsions” that not only proved difficult to treat, but also took away his son’s “intellectual vivacity and power to move his limbs”. His son did not only have epilepsy, he had a condition that both resulted in epilepsy and developmental regression. If the EEG had already been around at the time, it would have been very likely that his son would have presented with the EEG pattern of hypsarrhythmia. A condition where epileptic activity is so prominent that it affects cognition has become known as epileptic encephalopathy and the use of this concept is the topic of a recent commentary by Howell, Harvey, and Archer in Epilepsia.
State, not syndrome. Reading the review by Elling and collaborators on the historical perspective on West Syndrome, I kept wondering whether Dr. West’s son had infantile spasms that eventually resolved or whether he had a genetic epilepsy that presented with Infantile Spasms. The recent commentary by Howell, Harvey, and Archer reviews our current understanding and use of the concept of epileptic encephalopathy in detail, rightfully criticizing that we often confuse the concept of epileptic encephalopathy with a syndromic diagnosis. In fact, epileptic encephalopathy refers to a state rather than a diagnosis. A patient with epilepsy may be in the state of epileptic encephalopathy at some point during the course of the epilepsy, but the encephalopathy itself is different from the overall diagnosis. The epileptic encephalopathy is the potentially reversible state that a patient with epilepsy can be in, but it does not represent the overall diagnosis.
Dravet Syndrome. To further explain this important distinction that Howell et al. point out: Dravet Syndrome may have excessive epileptiform activity at times that contribute to the cognitive difficulties, but the developmental plateauing that it is often seen in the second year of life appears to be independent of the epileptiform activity. Therefore, according to Howell et al., Dravet Syndrome is not an epileptic encephalopathy. Rather, patients with Dravet Syndrome can have epileptic encephalopathy as a component of their epilepsy. However, there can be patients with Dravet Syndrome and other patients with SCN1A-related epilepsies who never have an epileptic encephalopathy. To point out the extremes, there are patients with recurrent relentless seizures occurring every few minutes who do not have impairment of their mental status. By definition, these patients do not have an epileptic encephalopathy even though they basically have constant seizures.
On purpose. While writing this blog post, I am thinking how best to explain why the epilepsy genetics field has systematically ignored this distinction of diagnosis and functional state. On purpose, the epilepsy genetics community has used epileptic encephalopathy as a diagnostic category to provide a broad label for patients with severe epilepsies. It is the only thing that is am somewhat missing from the review by Howell, Harvey, and Archer: I would have wished that the authors had tried to elucidate why this term is frequently misused. In fact, I believe that using epileptic encephalopathy as diagnostic category has been incredibly useful for the community. Here are three reason that I think that using the concept of epileptic encephalopathies as a diagnostic category is meaningful.
1 – Finding causes. Grouping patients under a broad diagnostic label has been extremely helpful to identify the underlying cause of disease. While traditional epileptology provides us with specific syndromes such as West Syndrome, Lennox-Gastaut Syndrome, or Dravet Syndrome, there is no single term that encapsulates all patients with these conditions. We may call them severe epilepsies of childhood, rare epilepsy syndromes, or early-onset catastrophic seizure disorders, and we have used many of these concepts in the past. For some reason, “epileptic encephalopathies” was the term that stuck and was felt to be most appropriate, indicating that severe epilepsy and cognitive issues are the main characteristics in this broad patient group. There are various reasons to capture a broad range of patients. In the genetics world, we are used to surprises. Inclusion of unusual patients has often resulted in findings that we did not expect to start with. This is how we found 15q13.3 microdeletions, CHD2, KCNA2, STX1B, HCN1, GRIN2A and a list of at least 10 other genes. If it had not been for a unifying term rooted in traditional epileptology, it would have been difficult to allow for the merger of genetics and epileptology that we have seen in the last decade – the explosion of gene discovery that is laying the ground for precision medicine in these conditions. I would see the term “epileptic encephalopathy” as call to action to move forward, recycling an old epileptological concept to provide the basis of discovering the causes of diseases that have a joint clinical presentation.
2 – Impossible to disentangle. I am aware why it makes sense to restrict the concept of epileptic encephalopathies to aspects of the disease where excessive epileptiform activity contributes to cognitive issues. It is the most straightforward thing to do from a clinical perspective when focusing on the epilepsy alone. However, I would argue that in many genetic epilepsies, the individual contribution of epileptic activity and underlying genetic alteration may be difficult to disentangle. Only a subset of epileptic encephalopathies are genetic, but these conditions may provide the best example on how complex the interplay of seizures, EEG, and cognition may get in clinical practice, especially in children with pre-existing disabilities. In many cases, there are other causes of regression, stagnation, or plateauing than the epileptiform activity, but exactly quantifying the contribution of epileptiform activity to the overall cognitive deficits may be difficult. Children with specific genetic epilepsies can have cognitive issues not only with obvious EEG activity, but also with frequent, self-limited seizures in the same way that they are often more susceptible to encephalopathy with infections that would be expected from the EEG alone. After a cluster of seizures, some patients with genetic epilepsies can have prolonged episodes of regression with normal EEG findings. This is in contrast to the exemplary patients with recurrent relentless seizures that I have mentioned above. I am not saying that I am having a perfect answer for this, but I feel that there are many aspects in the disease course of patients with genetic epileptic encephalopathies that we currently do not understand. My concern would be that we would be blind to these aspects of the disease if we limit ourselves to epileptic encephalopathies in a strict sense.
3 – Empowering the rare disease community. As mentioned above, using the term epileptic encephalopathies (“the EEs”) as a diagnosis has made it possible for the professional community to come together by providing a suitable construct that allows epileptologists and geneticists to find common grounds. It has made it possible to transform a group of rare neurological diseases into genetically identified etiologies, which has spurred a huge activity of patient initiatives and medication trials in rare epilepsies. Basically, it has made the rare epilepsies so prominent that they are a valuable target for biotech to invest in. The Cinderella of medicine (W.G. Lennox) has become a prominent member of the rare disease community. I feel that losing the precision of the correct concept of epileptic encephalopathy is a price worth paying for this. Again, think of “epileptic encephalopathy” as concept that provides us sufficient momentum to move forward, a concept that suggests, but not proves, that there is some aspect of the disease that we might want to treat.
Summary. I have tried to explain why the epileptic encephalopathies have emerged as a diagnostic category rather than a description of the potentially reversible state that a patient may be in at a given time. While I agree that it is important to have a concept that reflects the actual state when epileptic activity impacts on cognition, language, and behavior, the epilepsy genetics community has coalesced around epileptic encephalopathies as a diagnostic category. Countering the argument by Howell, Harvey, and Archer that we should come up with a new term for the diagnostic categories, I would argue that by 2016 the term epileptic encephalopathy may be firm in the hands of the epilepsy genetics and rare disease community.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.

This is fascinating information to a mother with a son dx with West Syndrome and has gone on to have now dx with LgS and the word enciphilitis is all over his chart.
What, if anything have you heard about a gene mutation on the UBE2A gene? My son presented earlier with seizure-like episodes and EEG shows LGS-like patterns of spike and wave while his body is not having a physical seizure. When they did manifest in a manner I could see – they looked like he was staring off into space, or his entire body would be rigid while doing a terrible scream.
I’ve found the UBE2A gene has a symptom of seizures, but many of the people in the minimal and previous case studies never had MRIs. My Son’s MRI shows that his hippocampus are slightly atrophied/rotating, large pineal cyst and grey matter in the white matter though out both hemispheres.
He hasn’t had any physical manifestations of seizure-like episodes since we started CBD in June of 2015 and a small dose of Klonopin (That we have weaned by 3/4, but his EEGs remain with the LGS spike/wave through-out the day and night.
We have yet to find someone who can explain this to us/what it means. Can you help?
Dear Erin,
unfortunately, we know very little about the epilepsy in patients with UBE2A pathogenic variants. My understanding is that loss-of-function mutations are associated with a particular form of X-linked intellectual disability referred to as Nascimento-type intellectual disability. Some patients are known to have seizures, but I am unaware of any study that looks at the seizures in more detail.
Best wishes,
Ingo