
Haploinsufficiency. STXBP1-related disorders are one of the most common neurodevelopmental disorders due to pathogenic variants in a single gene. Haploinsufficiency is the proposed disease mechanism and a significant number of individuals have deletions or protein-truncating variants. However, there are also recurrent missense variants in STXBP1, which is often seen in diseases that have a different disease mechanism. In a recent publication in Nature Communications, some of the recurrent variants in STXBP1 are suggested to have an additional disease mechanism, a dominant-negative effect. In this blog post, I want to discuss how we can reconcile both observations and whether STXBP1-related disorders are a single entity with a common disease mechanism.
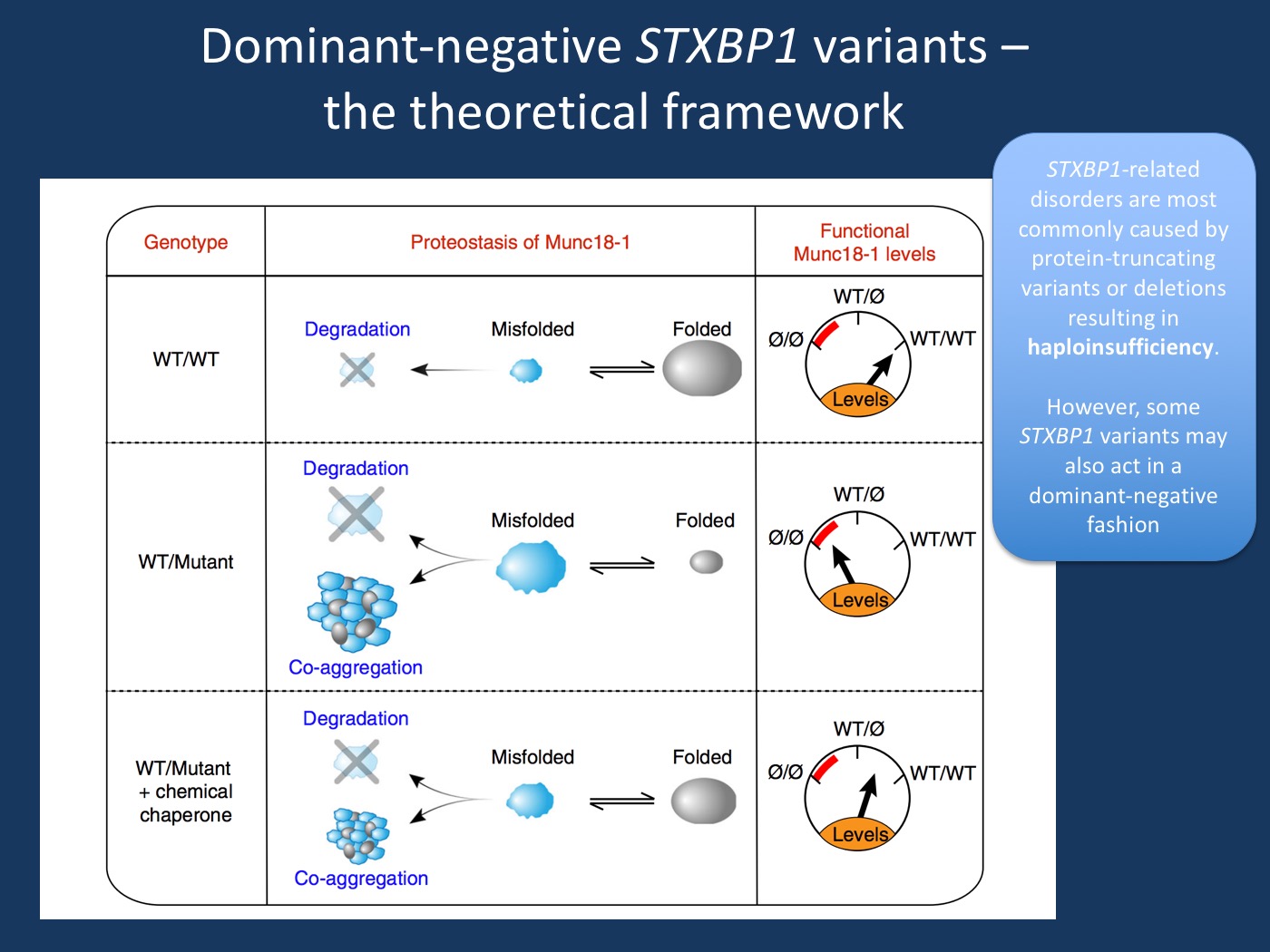
Figure. Model of Munc18-1 dysfunction due to a dominant-negative disease mechanism in encephalopathies and the potential rescue of deficits with chemical chaperones [modified under a Creative Commons Attribution 4.0 International License from Guiberson et al., 2018 https://www.nature.com/articles/s41467-018-06507-4]. Munc18-1-linked encephalopathies are caused by a dominant-negative disease mechanism. In the study by Guiberson et al., 2018, heterozygous missense mutations in Munc18-1 cause a reduction in functional Munc18-1 levels below 50%, due to a variety of mechanisms, including accelerated degradation of mutant Munc18-1, aggregation of misfolded mutant Munc18-1, and co-aggregation with wildtype Munc18-1. Chemical chaperones shift the unfolded–folded protein ratio toward a folded state, and result in an increase in total Munc18-1 levels. In the study by Guiberson et al., 2018, this overall increase in Munc18-1 levels is sufficient to rescue the Munc18- 1-linked neuronal deficits. In summary, even though haploinsufficiency is the established disease mechanism in STXBP1-related neurodevelopmental disorders, some missense variants appear to have a dominant negative effect in the model systems that Guiberson and collaborators used.
STXBP1. The STXBP1 gene encodes the Munc-18 protein, a critical component of the presynaptic machinery that is required for synaptic vesicle release. By interacting with both vesicle- associated proteins such as synaptobrevin 2 or VAMP2 and membrane-associated proteins such as Syntaxin-1 and SNAP25, the STXBP1 protein is a potent modulator of vesicle fusion. In fact, presence of STXBP1 is critical for vesicle fusion to occur, as absence of STXBP1 leads to the complete absence of synaptic transmission. The somewhat complicated name Syntaxin-binding protein 1 (STXBP1) therefore betrays the important role that STXBP1 has in making sure that synaptic vesicle fusion occurs in a regulated manner.
STXBP1 and disease. STXBP1 was first identified as a gene for human disease when deletions of the STXBP1 gene were identified in patients with Ohtahara Syndrome. Since this initial description, the phenotypic spectrum has widened significantly, encompassing a wide range or neurodevelopmental disorders ranging from isolated intellectual disability to severe, early-onset epilepsy. The reason why the clinical presentation of patients with STXBP1 is variable is unknown. There does not seem to a be genotype-phenotype correlation and patients with the identical pathogenic variant can have a range of clinical presentations. Phenotypic heterogeneity in neurodevelopmental disorders is a common scenario and even though STXBP1-related disorders have a wider phenotypic range than most genetic disorders, a wide range of clinical phenotypes is a typical feature seen in neurodevelopmental disorders. Disease-causing variants in STXBP1 are typically de novo and a significant proportion of variants in STXBP1 lead to haploinsufficiency, i.e. the loss of one functional copy of the STXBP1 gene. This includes whole gene deletions, partial gene deletions, or protein-truncating variants that including stop variants, frameshift variants, or splice-site variants. However, there are also many missense variants where the functional consequence cannot easily be determined from the variant itself.
Recurrent variants. Recurrent missense variants in STXBP1 are rare and most disease-causing variants are distributed throughout the gene – a typical findings for disease with haploinsufficiency as the disease mechanism. However, some patients carry recurrent missense variants. Some of these variants are amongst the variants examined by Guiberson and collaborators. When we reviewed the phenotypes of STXBP1-related neurodevelopmental disorders in 2016, we paid particular attention to some of the recurrent missense variants. We found that seven patients with the most frequent recurrent mutation p.Arg406His had a relatively uniform disease phenotype with seizure onset in the first months of life with severe or profound intellectual disability. In contrast, epilepsy course and cognitive outcome was more variable for other recurrent mutations such as p.Arg292Cys, p.Arg292His and p.Arg551Cys. Therefore, at least a single recurrent variant p.Arg406His may clinically be distinct – homogeneous rather than a spectrum – to postulate a disease mechanism other than haploinsufficiency that is overwhelmingly supported by the genetic architecture of the STXBP1-related neurodevelopmental disorder.
Study by Guiberson and collaborators. In their recent publication in Nature Communication, Guiberson and collaborators examine the functional consequences of several STXBP1 missense variants in a variety of model systems including yeast, C. elegans, and conditional knock-out mice. In particular, they look at the p.P335L, p.R406H, p.P480L, p. G544D, and p.G544V missense variants that overlap with the recurrent missense variants seen in patients. The authors find that these missense variants aggregate and bind to wildtype Munc18-1 (the STXBP1 protein) and result in a functional depletion that is lower than what would be expected by haploinsufficiency alone. This is called a dominant-negative effect, a phenomenon known from other genetic etiologies for epilepsy such as KCNQ2 encephalopathy. Basically, the variant protein is not only non-functional, it also affects the non-variant copy. The authors continue to show that chemical chaperones that can affect protein structure can alter some of the function effects that is caused by the apparent dominant negative effect.
Haploinsufficiency or dominant-negative.The publication by Guiberson and collaborators raises an interesting hypothesis even though this is not spelled out by the authors directly: STXBP1-disorders do not have a uniform disease mechanism, but may have different disease mechanisms depending on the variant. While haploinsufficiency will remain the prominent disease mechanism, there may be the possibility that at least some of the recurrent missense variants, most prominently the frequent p.R406H variant may do more than disrupt one copy of the gene – they may act in a dominant negative manner. Two different disease mechanisms are not unusual. This has been shown in KCNQ2 and has been suggested in SCN2A. Therefore, hypothesizing that at least some patients with STXBP1-related disorders have variants that lead to a dominant-negative effect is an interesting hypothesis that may help identify different treatment strategies for a subset of patients.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.

Very interesting! KCNA2 has three functional classes: gain- and/or loss-of-function.