
Trio exomes. The concept of neurodevelopmental disorders is an umbrella term including intellectual disability, developmental delay, and autism spectrum disorder. About one quarter of these patients have epilepsy, including epileptic encephalopathy, in which the epileptic activity itself contributes to developmental delay or regression. One major cause of these disorders are de novomutations, which are present in the child but not present in either of the parents. A recent publication in Nature Genetics looked for de novo variants in nearly 6,700 patients with neurodevelopmental disorders, nearly 2,000 of whom had epilepsy. This study is an order of magnitude larger than the largest previous study of this kind and represents an important effort in epilepsy genetics. Here is what we want to review their major findings.
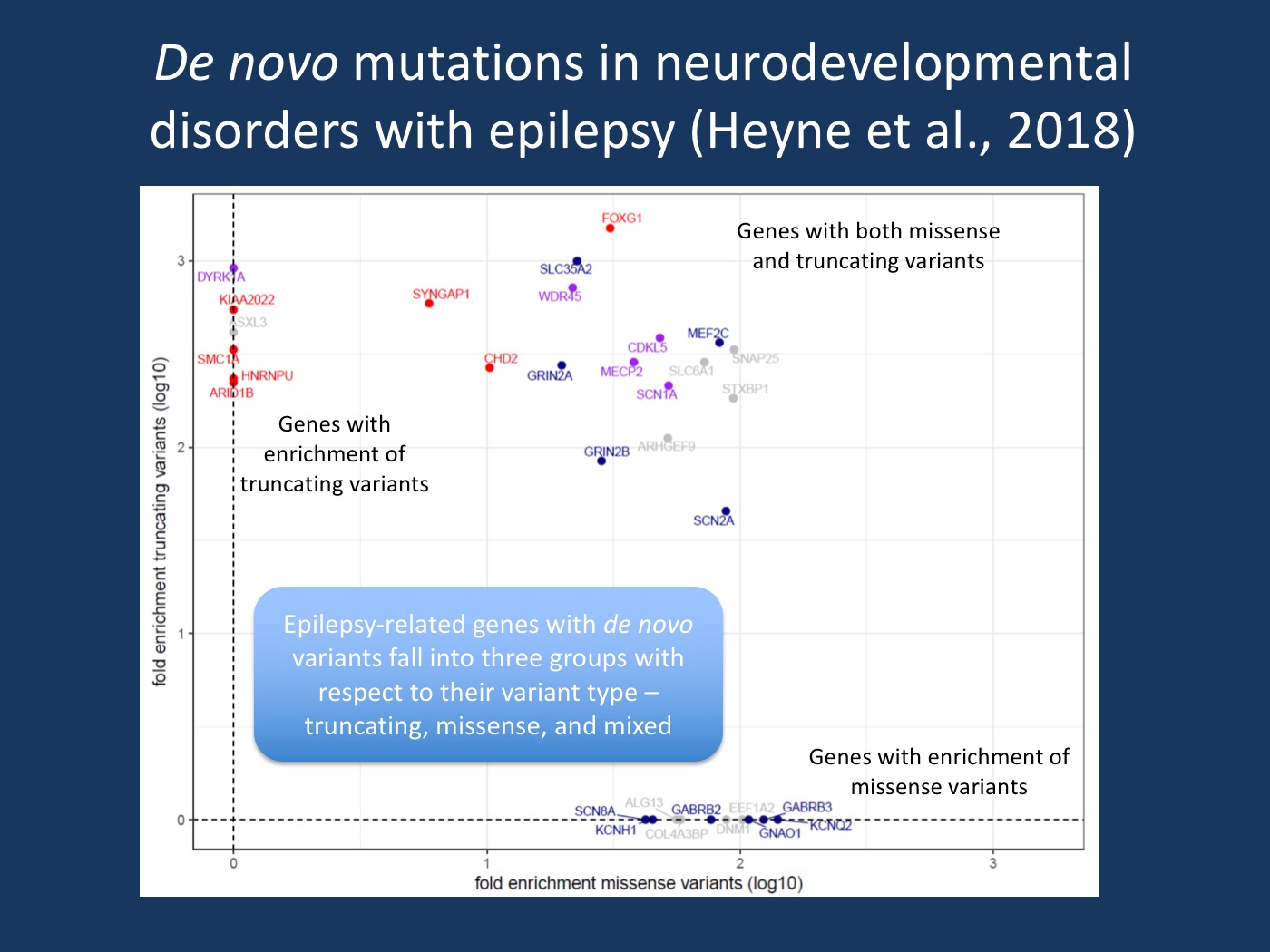
Missense versus truncating DNV per gene. Plot of fold enrichment of missense versus truncating de novo variants of 33 genes enriched in NDD with epilepsy. Genes are colored after the consequence of likely pathogenic or pathogenic de novo variants reported in ClinVar. Genes with only missense DNV reported in ClinVar are colored blue, only truncating red. Genes with both missense and truncating de novo variants in ClinVar are colored purple, genes without DNV in ClinVar grey (=unknown). [Modified Supplementary Figure S2 from the bioxriv version of the manuscript by Heyne and collaborators (https://doi.org/10.1101/123323), permission granted by the authors].
GABRB2. This gene encodes the β2 subunit of the gamma-aminobutyric acid (GABA-A) receptor. Five epilepsy patients in this cohort had missense mutations in GABRB2, in addition to two cases with epilepsy and intellectual disability which were previously reported in the literature. Functional studies of one of these previously published mutations found decreased function of the GABA-A receptors, consistent with a true pathogenic role this gene. The phenotype associated with this gene is not yet clear, and one patient in this study had NDD without epilepsy.
SNAP25. This gene encodes a protein in the SNARE complex which plays a role in neurotransmitter release at synapses. Four patients in the study had de novo variants in this gene, all of whom had epilepsy. These mutations all occurred an important functional domain of the protein, a region which is highly intolerant of variation. Mice with mutations in SNAP25 have seizures and cognitive dysfunction. The phenotypic spectrum of this gene is also not yet clear. One previous published case had congenital myasthenia, cortical hyperexcitability, cerebellar ataxia, and ID.
Which genes and which variants? Most of the genes implicated in patients with epilepsy were also implicated in patients with neurodevelopmental disorders without epilepsy. This is not surprising given what we have learned about the overlapping genetic determinants of neurocognitive disorders and the phenotypic variability associated with these genes. Were any genetic findings specific to epilepsy, as opposed to neurodevelopmental disorders without epilepsy? Mutations in ion channel genes were more common in patients with epilepsy. More surprising was the finding that missense variants, as opposed to truncating variants, were more common in patients with epilepsy, and this was true in both ion channel genes and genes encoding other kinds of proteins. One might expect truncating variants to have more severe consequences than missense variants. But it was not the case that the epilepsy patients were on the milder end of the neurocognitive spectrum—in fact, the opposite was true. One possibility suggested by the authors is that epilepsy involves altered protein function more often than haploinsufficiency, although both kinds of functional effects are known to cause epilepsy. For example, a gain-of-function effect as known in SCN2A or SCN8A may be more likely to result in epilepsy compared to a loss-of-function variant. The cause of the curious finding remains to be determined.
Therapeutic relevance. Of the 33 epilepsy genes identified in patients included in this study, five genes have well established treatment implications. In total, these five genes accounted for 28% of the de novo variants in found in epilepsy genes, and 5% of all the qualifying de novo variants detected by the study. These numbers highlight the growing impact of genetic diagnosis on the treatment of patients with epilepsy and neurodevelopmental disorders, which will continue to grow as new precision treatments based on specific genes and specific variants become more commonplace in the future.
What you need to know. In summary, the publication by Heyne and collaborators provides a solid statistical basis to our current understand of what constitutes an epilepsy gene. Most of the genes suggested in this largest meta-analysis so far were previously known, but the study also showed that new genes such as GABRB2 and SNAP25 are still out there, requiring large sample sizes. Most importantly, however, the study by Heyne and collaborators is the first study that tries to separate the epilepsy-specific effect within large genetic cohort studies. Phenotyping in these studies is often limited, but the authors still come up with provocative results, such as the higher frequency of missense mutations. Future studies like this will help us understand the role of epilepsy in the larger framework of neurodevelopmental disorders.