
Controversy. Our recent post that featured our Neurology publication on STXBP1 generated much interest, discussion, and debate. In particular, we received feedback from the online STXBP1 parent community that our assessment of STXBP1 encephalopathy as a static rather than a degenerative disease may be incomplete. Let me try to reconcile the results of our study with the experiences that families have shared with us in the last two weeks, trying to understand how STXBP1 can be a disease with many faces and what the common features are.
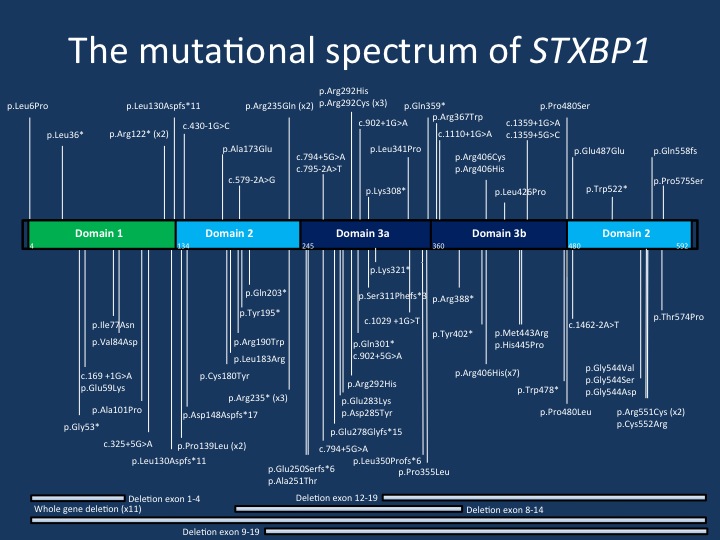
The mutational spectrum of STXBP1. The current figure displays the majority of the mutations reported in the publication by Stamberger and collaborators. The mutations are spread across the STXBP1 gene, but there are a few mutational hotspots. The p.Arg406His mutation is the most common recurrent de novo mutation in the STXBP1 gene.
Neurology meets Facebook. Let me start out that we appreciate the time everyone took to share their thoughts and experiences regarding STXBP1. What we are trying to do in this post to put both the results of our study and the experience of the online patient community into context. To summarize the current controversy: in our study of 147 patients with STXBP1 encephalopathy, we found very little evidence of regression in patients with STXBP1. To be precise, 4/147 patients had some degree of regression. We were challenged by the STXBP1 online parent community that this maybe an underestimate of the true frequency of regression in patients with STXBP1. Even though the Channelopathist is not a platform for sharing individual patient information or to give or receive medical advice, we appreciate input that pertains to the larger picture of a disease. What I am trying to do in this post is to address the question regarding STXBP1 related regression scientifically with the data that we have.
Limitations. Our Neurology study did not follow individual patients over long periods of time to track their outcomes. The data are not population based. But it is an important first attempt at summarizing everything we know right now about STXBP1, in 2016. At this point this is the most comprehensive view we have of STXBP1 encephalopathy, reporting the largest cohort of patients described to date, referred from clinicians all over the world. But it is important to keep in mind that this is the 10,000 foot view – the sum of nearly 150 patients in total. What families are living with every day is the upclose view from the frontlines – the realities and struggles that can perhaps never accurately be captured in a scientific publication. Without a doubt, our understanding of STXBP1 encephalopathy will continue to evolve, and it is always valuable to get feedback from families of patients who understand the disorder in ways that researchers and clinicians never truly can. As scientists, we need to keep an open mind that maybe some of what we think we understand today may need to be revised tomorrow.
Framing the question. Prior to asking questions about the frequency of regression in patients with STXBP1, let me put the scope of our study into context. Our study was not intended to go into the details of the developmental regression in the patients who were included, but was focused on the overview of the disease. Again, while these type of studies never have the granularity of a longitudinal assessment of a single patient, they offer some insight into the larger landscape of the disease phenotype from the collective experience of the epilepsy community. Rephrasing the question that we were asked, the main issue is how frequently patients with STXBP1 encephalopathy have unexplained loss of milestones that is not explained by other factors.
1 – Cause of the regression. STXBP1 encephalopathy is not a classic neurodegenerative disorder like the neuronal ceroid lipofuscinoses (NCL) or leukodystrophies. As far as we understand, there is no inherent disease progress that would result in progressive neuronal death or organ failure. However, patients with neurodevelopmental disorders may be particularly susceptible to the effect of severe infections or other systemic disorders. A prominent example of this are mitochondrial or neurometabolic disorders, where patients may have crises and may accrue non-reversible brain injury during these episodes. STXBP1 encephalopathy is not known to be a disease of energy failure or a condition where sudden metabolic derangements occur. There are some isolated reports about mitochondrial dysfunctions, but this issue has not been assessed systematically and is difficult to reconcile with what we know about the function of the Munc18 protein. Therefore, in future studies assessing regression in patients with STXBP1 encephalopathy we will need to pay particular attention to the circumstances under which the loss of skills or regression occurred.
2 – Patient population. STXBP1 encephalopathy is not a homogeneous disease and one question that researchers typically ask when assessing a phenomenon is whether all patients with this phenomenon actually had the particular disease in question. For example, some patients with STXBP1 encephalopathy may have larger genomic rearrangements deleting STXBP1 in addition to other contributing genes. Other patients may have additional disorders that increase the risk for regression. The question in these studies is to define the patient population, such as trying to assess the frequency of regression in patients with pathogenic de novo STXBP1 variants who had other contributing causes sufficiently excluded. We would usually assume that STXBP1 variants are so rare that the distinction between pathogenic and non-pathogenic variants is easy. However, given a frequency of missense mutations of up to 40%, it may not be easy to tell pathogenic variation from non-contributory, inherited variants.
3 – Recruitment bias. All studies have a recruitment bias and it is important to be aware how patients were ascertained. In our study, patients were largely ascertained from the literature and from referring clinicians. As systematic testing is mainly performed in younger patients, this results in a median age of inclusion of ~6 years. This cohort is different from patients who may present to a genetics clinic with long-standing disease of unknown origin that is finally identified to be STXBP1. Both ascertainment methods capture a certain aspect of the disease and the conditions may look entirely different. In order to reconcile both view, we need some estimate on how STXBP1 encephalopathy looks like in the entire population.
4 – Population frequency. In our study, we used data from our Danish collaborators. The big advantage of the Danish cohort is that all patient with epilepsy are seen in a single center. As the frequency of STXBP1 encephalopathy is small, the total number of patients is also small. There were 13 Danish children born between 1987 and 2012 who were referred to the Danish Epilepsy Centre and diagnosed with STXBP1 encephalopathy. This data, despite the small numbers, is our best estimate at population-based data that we have at this point. Reviewing the Danish data with one of the Principle Investigators in the last week suggested that the median age of the patients is around 15 years with 5 patients above the age of 18. None of the patients had phenomena consistent with regression. While this small number does not exclude that a certain percentage of patients may have regression, it emphasizes that regression is not a consistent feature of this disease and does not occur in the majority of patients.
5 – Counseling. The discussion about STXBP1-related neurodegeneration relates to counseling and giving families a long-term perspective with regards to a certain disease. Please be aware that it is not our role on the Channelopathist to provide counseling or to provide data that will be used for counseling. This will always be the domain of a discussion with a qualified provider who knows the patients and all the relevant information. With regards to the overall landscape of STXBP1 encephalopathy, I think that both the experience of the parent community and our study do not contradict each other. STXBP1 is a neurodevelopmental disease with many facets and depending on the ascertainment of patients, some features are more prominent in a specific patient population. With regards to the risk for regression in a patients with STXBP1 encephalopathy, we can only provide rough estimates on the frequency that our study may be missing. We can be certain that this phenomenon does not occur in most patients, but a comprehensive assessment of adult patients with STXBP1 is currently not available.
A preliminary conclusion. There is controversy in the patient community regarding regression in patients with STXBP1 encephalopathy. I have outlined a few aspects that require further research and clarification regarding this phenomenon. Data from our study and including population-based data suggests that regression in patients with STXBP1 encephalopathy may occur, but is rare, even in patients who are followed up into adolescence. In patients with STXBP1 encephalopathy and regression, future studies will need to look deeper into the implicated genetic changes in the STXBP1 gene, other contributory factors, potential modifying factors and triggers for the regression. Also, we will need a better understanding of the adult STXBP1 phenotype.
Moving forward. Finally, I would like to apologize for not replying to many of the comments that we received regarding STXBP1. We are in the middle of creating the framework for our registry and hope that we can address some of these questions once we have a formal framework for this. Building a registry is not fast – it needs to be thorough to make sure that we capture the data that we need. We’re looking forward to working closely with the STXBP1 community to establish a formal family organization and patient registry and working alongside the patients and families to further our understanding of STXBP1 encephalopathy. As you can tell, we have not made many of the comments on this blog public. If you’d like to share your opinion publicly, please indicate whether this is ok with you. If so, we would ask you not to present identifiable patient data and respect our rule that the Channelopathist is a scientific community resource for academic discussion and not a resource to provide, receive, or exchange medical advice.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.

I’d just like to share for families looking for support we have a closed group on Facebook
Parents of kids with STXBP1
You can find us in FB search.
This should be the url
https://www.facebook.com/groups/123917951055617
Thank you! As soon as our registry is live, we hope that we can formally connect with the STXBP1 families and assess some of the issues that families have reported.
Thank you Dr Helbig for all your hard work!