
Synaptic. This is STXBP1 week and things are currently happening in rapid succession. We are getting ready for the first STXBP1 Charity Ball and our publication in Neurology reviewing the phenotypic features of 147 patients recently came online. STXBP1 is one of the five most common genes for epileptic encephalopathies and related neurodevelopmental disorders. However, in contrast to SCN1A, SCN2A, CDKL5, or SCN8A, it has received relatively little attention in the past from the epilepsy community. Let’s revisit a common epilepsy gene that holds more secrets than most people would imagine.
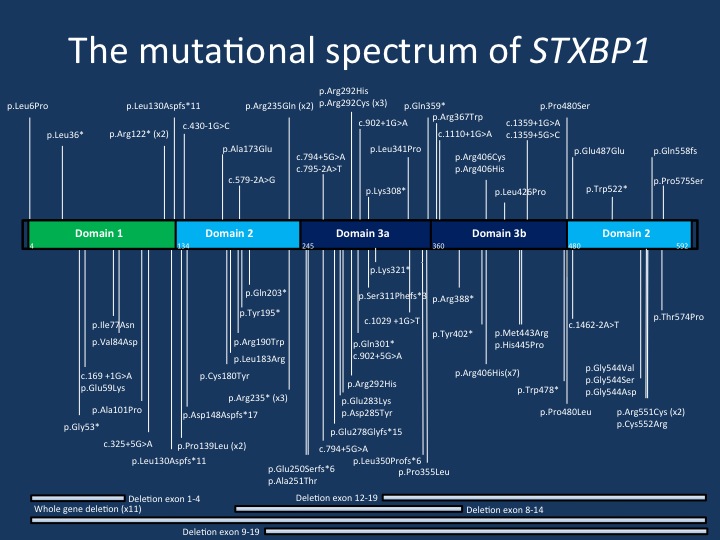
The mutational spectrum of STXBP1. The current figure displays the majority of the mutations reported in the publication by Stamberger and collaborators. The mutations are spread across the STXBP1 gene, but there are a few mutational hotspots. The p.Arg406His mutation is the most common recurrent de novo mutation in the STXBP1 gene.
Stick it to STXBP1. Prior to a discussion of some the findings in our recent publication, let me send you a last minute invitation to the STXBP1 Charity Ball on February 20th at the Doylestown Country Club. During AES 2015 in Philadelphia, we had our first STXBP1 parent night at CHOP and we are currently in the process of building an international STXBP1 registry. Therefore, it is timely that our first larger STXBP1 publication appears this week, providing us with a big picture snapshot of the overall STXBP1 landscape. For this publication, spearheaded by Hannah Stamberger as the first author and Sarah Weckhuysen as the senior author, we extracted data on published patients from the literature and added phenotypic features of 45 previously unreported patients.
The STXBP1 phenotype. STXBP1 was historically considered the gene for Ohtahara Syndome as deletions in this gene were first discovered in this phenotype. The STXBP1 gene codes for an essential subunit of the synaptic fusion machinery that enables synaptic transmission. This gene, which also goes by the synonym Munc-18, is required for synaptic transmission – mice in which the Munc-18 gene is deleted have no synaptic transmission even though the structure of the Central Nervous System develops normally. The genetic mechanism in STXBP1 encephalopathy is haploinsufficiency, i.e. only one functional copy of the STXBP1 gene remains present in patients with STXBP1 encephalopathy, as the other copy does not produce a functional protein due to a deletion or mutation. Why this haploinsufficient state results in a severe neurodevelopmental disease with epilepsy remains unclear. It is assumed that interneurons are more affected by this haploinsufficiency than excitatory neurons, which then leads to net hyperexcitability. The clinical picture of STXBP1 encephalopathy has always been confusing to the epilepsy community. While some patients have well-defined epilepsy syndromes such as Ohtahara Syndrome or West Syndrome, most patients have an Early Onset Epileptic Encephalopathy (EOEE) that defies classical diagnostic criteria, sometimes with features of various epilepsy syndromes. Here are the five main messages of the publication by Stamberger et al that emphasizes the complexity of the STXBP1 phenotype.
1 – The two dimensions. I have focused on the two dimensions of STXBP1 in the title of this blog post. Basically, this concept refers to the fact that even though most patients in our series have severe intellectual disability, there is very little correlation between seizure onset, seizure severity, and the degree of intellectual disability. This is somewhat astonishing from a clinical perspective. In some patients with STXBP1 encephalopathy, the epilepsy already starts in the neonatal period. However, the neurodevelopmental outcome in these patients is not significantly different from patients who have seizures starting at a later age. In our analysis, we find no correlation between the “epilepsy dimension” and the “neurodevelopmental” dimension.
2 – Dynamics of the seizure phenotype. Epileptic spasms, focal seizures, and tonic seizures are the main seizure types and most patients with STXBP1 encephalopathy have multifocal epileptiform activity and hypsarrhythmia. Therefore, the combination of epileptic spasms and hypsarrhythmia appears to be the core phenotype of STXBP1 encephalopathy, even though there is considerable variation around this theme. Particularly some of the focal seizures and multifocal EEG findings are often a source of confusion in clinical practice and may suggest an underlying focal cause of the epilepsy in the initial work-up. The seizures in STXBP1 encephalopathy are highly dynamic and start early with a median onset of 6 weeks, presenting with spasms in more than 60% of cases. However, a small subset of patients have other distinct epilepsy syndromes such as Dravet Syndrome in 2% of patients. In contrast to many other genetic epilepsies, more than 40% of patients with STXBP1 encephalopathy become seizure-free. This is in stark contrast to Dravet Syndrome with mutation in SCN1A, CDKL5 encephalopathy, or DNM1 encephalopathy.
3 – Other neurological features. There are several uncommon features reported in some patients with STXBP1 encephalopathy including juvenile Parkinsonism, which prompted us to look at other neurological features provided that this information was available. We found that predominantly axial hypotonia, ataxia, tremor, spasticity, and dystonia were reported features in many patients. It is difficult to provide precise frequencies for each feature. We also addressed the question whether regression is a common feature in STXBP1 encephalopathy, a common clinical question that arises when talking about prognosis with families. We only found a very small subset of patients where regression has been reported. As far as we can tell in 2016, STXBP1 is not a neurodegenerative disease, but a static encephalopathy.
4 – Genotype/phenotype and recurrent mutations. In total, we were able to summarize 123 different STXBP1 de novo mutations, with most mutations only seen in a single individual. This provides a heterogeneous mutational landscape that is typically seen in conditions that result from haploinsufficiency. Overall, there was no association between mutation type and degree of intellectual disability or seizure outcome. This indicates that there is no obvious genotype/phenotype correlation for STXBP1 encephalopathy. Furthermore, there are several mutational hotspots in STXBP1. We found 13 recurrent mutations in STXBP1, including p.R406H in seven patients and p.R551C in four patients. All seven patients with p.R406H had seizures starting at 2.5 months and had severe to profound intellectual disability. However, the range of seizure phenotypes and neurodevelopmental outcomes was more variable for the other recurrent mutations, including p.R292C, p.R292H, and p.R551C.
5 – Population frequency. How frequent is STXBP1 encephalopathy? Given the centralized care for epilepsy patients in Denmark, where virtually all children with severe epilepsies are seen in a single center, we are able to estimate population frequencies of rare genetic epilepsies. For example, the population frequency for Dravet Syndrome is estimated to be 1:22,000. In contrast to the relatively homogeneous phenotype of Dravet Syndrome, the estimate for STXBP1 encephalopathy is more difficult. Using the same methodology, we arrived at a population frequency of 1:91,000. This, however, does not take into account patients with mild seizures who may not have been tested and does not account for patients with STXBP1 who do not present with seizures.
This is what you need to know. The recent publication by Stamberger and collaborators is a major step forward in our understanding of STXBP1 encephalopathy. We now have a good overview of the key clinical features of patients presenting with epilepsy and understand that even though the epilepsy may start early and very dramatically, a significant proportion of patients will become seizure-free. Also, there is little correlation between seizure freedom and developmental outcome. Most patients with STXBP1 encephalopathy have severe intellectual disability in our series. However, regression and loss of skills over time is an uncommon feature. There is little genotype/phenotype correlation and patients with identical de novo mutations may have different phenotypes. Overall, this emphasizes that STXBP1 is more than just an epileptic encephalopathy. It is a complex neurodevelopmental condition that requires multidisciplinary care.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
