
No name. The speed of gene discovery in human epilepsies is sometimes so fast that genetics beats biology. Some genes are implicated in disease faster than our ability to name them. In a recent publication, we describe the epilepsy phenotype of an X-linked gene that is only known by an identifier that indicates how little we know about it: KIAA2022. In contrast to a phenotype in males that is mainly characterized by intellectual disability, de novo mutations in KIAA2022 in females results in intractable myoclonic epilepsy.
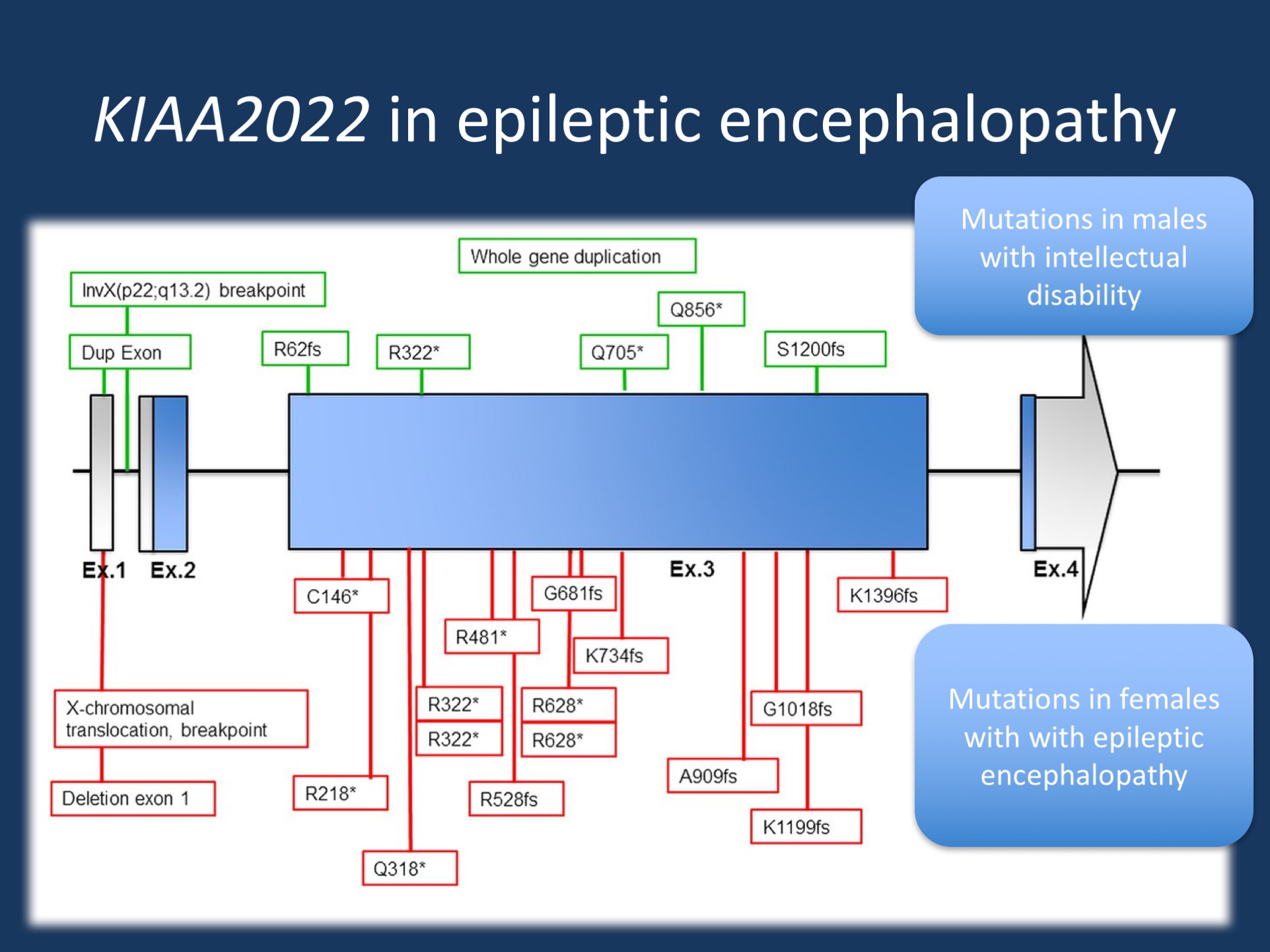
Location of mutations in KIAA2022 mutations in female patients (lower/red boxes) and previously reported mutations in male patients (upper/green boxes). Figure shows schematic presentation of known exon–intron organization of KIAA2022. Untranslated regions are indicated by grey color, and the coding regions are indicated by blue color. Adapted from the publication by de Lange and collaborators under a Creative Commons Attribution (CC BY 4.0) license.
From MAE to KIAA2022. In the recent publication by de Lange and collaborators, we describe the phenotypes of females carrying loss-of-function mutations in KIAA2022, a gene previously only known to cause intellectual disability in males. For the Kiel/Philadelphia team, the KIAA2022 story started with a EuroEPINOMICS-RES patient from our cohort of patient with Myoclonic-Astatic Epilepsy (MAE, Doose Syndrome). While several patients in this cohort did not have the classical phenotypes of MAE, the broader phenotype of patients included in this cohort turned out to be one of the most productive source of gene discovery for epileptic encephalopathies in the EuroEPINOMICS project.
The KIAA2022 phenotype. In the publication by de Lange and collaborators, we describe 14 female patients with KIAA2022 encephalopathy. 12/14 patients had epilepsy, which was mainly characterized by myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. In most patients with epilepsy, the seizures turned out to be refractory to multiple antiepileptic medications. Seizures usually started in infancy or early childhood, which may have given rise to the inclusion of KIAA2022 patients in a cohort of patients with MAE-like phenotypes. 13/14 girls with KIAA2022 encephalopathy had mild to severe intellectual disability. In 6/13 patients, the developmental delay preceded the onset of the epilepsy. 10 patients had prominent behavioral issues including autism, aggression, and hyperactivity. Given that some females only started with developmental issues after the onset of seizures in the first few years of life, this clinical observation reinforces the notion that the female KIAA2022 phenotype is not only an intellectual disability syndrome with mild epilepsy, but an epileptic encephalopathy where developmental stagnation or regression are correlated with the occurrence of seizures.
KIAA2022 in males. Mutations in the KIAA2022 gene have previously been identified as the cause of X-linked severe intellectual disability in males, sometimes with dysmorphic features. In males, there is little evidence that this gene is involved in epilepsy or seizures. Therefore, the finding that loss-of-function variants in KIAA2022 result in difficult-to-treat myoclonic epilepsy is surprising. Usually, with X-linked conditions, we would expect a milder phenotype in females, if any phenotype at all. The intellectual disability in females carrying KIAA2022 loss-of-function mutations fits this pattern, but the epilepsy is unusual. In our publication, we drew the parallel to PCDH19, a gene related to an epileptic encephalopathy in females where males carrying mutation are unaffected. In PCDH19, there is the hypothesis that the disease-causing effect is due to process called cellular interference, the mixture of neuronal cells with and without the PCDH19 protein during development. It can be speculated that the pathogenic effect of KIAA2022 may be due to a similar mechanism. This is relevant as there is very little known about KIAA2022 other some evidence that is may be expressed in neurites in the growing nervous system.
ExAC. Another twist in the KIAA2022 story was the nature of the loss-of-function variants in the ExAC database. Usually, for a gene related to a severe childhood-onset disease due to haploinsufficiency, we would not expect any loss-of-function variants in population databases. ExAC is not a pure control database, but it does not contain childhood-onset disorders. Therefore, the presence of KIAA2022 variants in ExAC is suspicious – and they turned about to be interesting examples on what we can see in public databases, ranging from variants escaping nonsense-mediated decay to sequencing artefacts. For example, a single truncating mutation (Arg1469*) is in the second to last exon. It is known that truncating mutations that distally in a gene can escape the cellular machinery that clears truncated mRNA transcripts, a mechanism called nonsense-mediated decay. It can be assumed that the Arg1469* mutation results in a shorted KIAA2022 protein that may still be functional. Another ExAC variant, the frameshift variant (Arg1085fs) appears to be a miscalled variant when looking at the trace files. In contrast, this variant may be an in-frame indel, a frequently seen, non-pathogenic genetic alteration. Identifying the ExAC variants as either distal truncation mutations escaping NMD or alignment artefacts further reinforces the idea that truncating mutations are not tolerated in healthy individuals, further adding to the suspicion that this gene is disease-causing.
What about carrier females? In families with affected males, carrier females are unaffected, and we believe that X-inactivation is playing a role. In our study we investigated X chromosome inactivation patterns in 7 patients, 6 of whom showed random X-inactivation patterns. Surprisingly one patient showed 100% skewing of X-inactivation, which correlated with a phenotype closely resembling the male phenotype of severe delay, hypotonia, microcephaly, dysmorphic features, and no epilepsy. A random pattern of X-inactivation appears to correlate with refractory myoclonic epilepsy without dysmorphic features. We proposed that X inactivation skewing in the other direction may account for the unaffected phenotype of carrier females.
This is what you need to know. In our recent publication, we describe the female phenotypes of loss-of-function variants in KIAA2022. This gene has been previously described in intellectual disability in males with severe intellectual disability and the newly described female phenotype has unusual features that would not be predicted by the phenotypic features seen in males. The female KIAA2022 phenotype is characterized by intractable myoclonic epilepsy starting in childhood, adding the KIAA2022 gene to the growing list of genes for epileptic encephalopathies.
Open Access. Finally, I would like to emphasize that the current KIAA2022 manuscript is an Open Access publication that is freely available to the epilepsy community. As we mentioned previously, the Channelopathist team feels strongly that Open Access publications are critical for the community as they provide a fair access to the research literature in the field.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
