
E2 consortium. Infantile Spasms and Lennox-Gastaut Syndrome are two epilepsy syndromes with a strong genetic component. De novo mutations play an important role in genetic epilepsies. However, given the overall mutational noise in the human genome, telling causative genes from innocent bystanders is difficult. In the largest and most comprehensive analysis so far, our E2 consortium just published a joint analysis of 356 patient-parent trios, which were analyzed by exome sequencing. In addition to implicating DNM1, GABBR2, FASN, and RYR3, this publication sends a clear message: the age of gene discovery in epilepsy is over – from now on, genes will find themselves. Let me tell you what I mean by this.
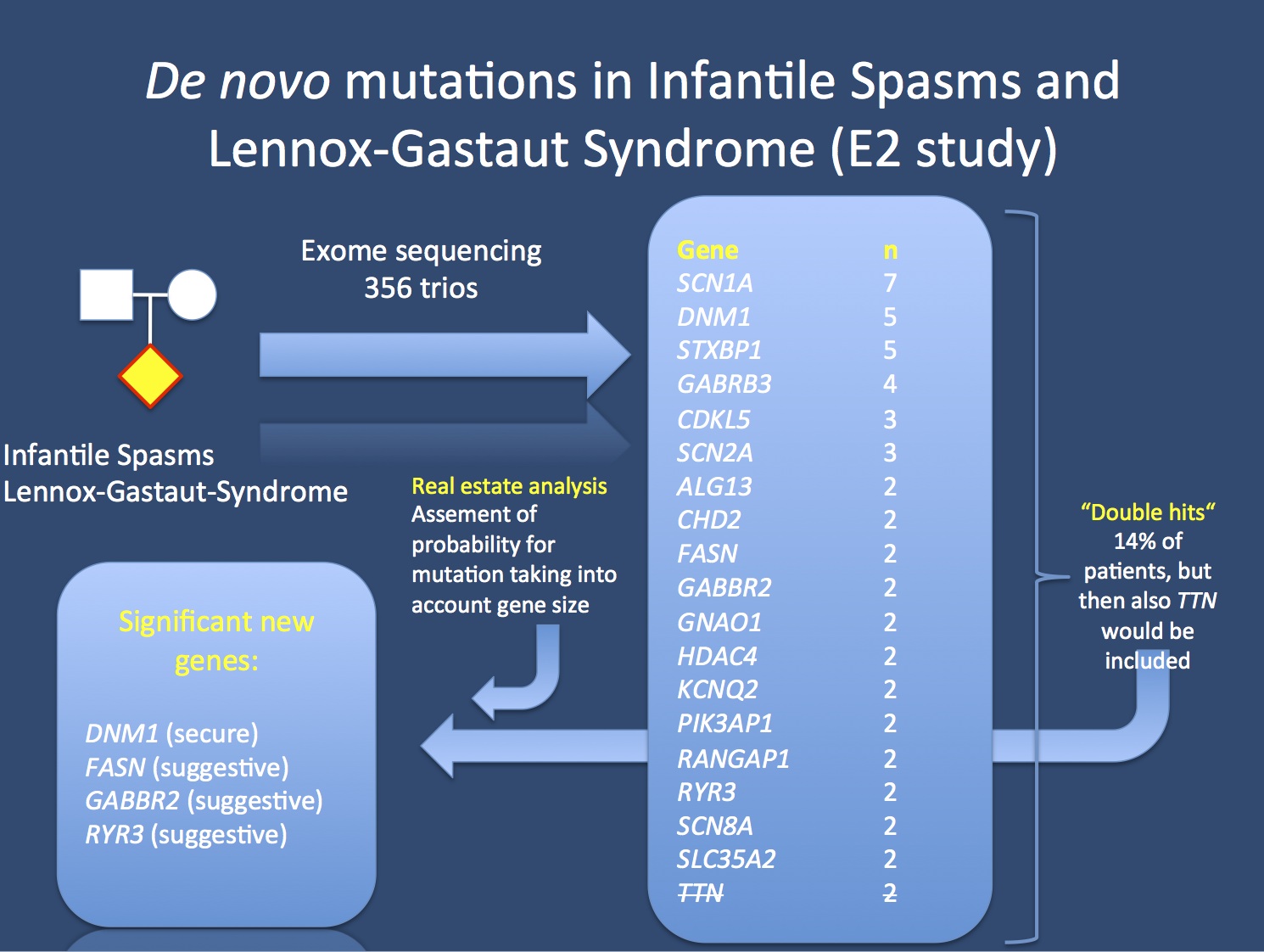
The E2 study analyzed 356 patient-parent trios with Infantile Spasms or Lennox-Gastaut Syndrome. In total, 12% of patients had genes that could be implicated as genes for epileptic encephalopathies. Some genes, however, even though they occurred in more than 1 patient, were not significant. TTN coding for titin (strikethrough) is one example, this gene is a known exome confounder. In order to tell causative genes from genomic noise, we used a real estate analysis that took into consideration the gene size.
De novo mutations. Just a brief reminder on how the field was only two years ago. Exome sequencing, initially announced as the new, wonderful tool in genetics, fully delivered its promise. It identified variant after variant in patients with neurodevelopmental disorders, but we soon realized that a similar river of rare variants can also be found in unaffected siblings. In fact, even in moderately large cohorts, the mutation rate in patients with autism was not higher than the mutation rate in their unaffected siblings. However, just as we were thinking about giving up or moving to other technologies, something amazing happened: we realized that using patient-parent trio sequencing in epilepsies managed to identify recurrent genes that withstood the test of time. While other fields of neurogenetics such as autism or schizophrenia genetics are still struggling to identify causative genes, the story is much different in epilepsy. Over the course of the last 24 months, we had securely implicated at least 8 novel genes. But the story wasn’t over yet.
Epi4K. The initial Epi4K publication was the launching pad for a series of collaborative efforts that ultimately led to the formation of the E2 consortium. Our basic question was: what would happen if we joined samples and increased the sample size? And here is the answer: by joining datasets and expanding the cohort to additional patient-parent trios, we can provide solid evidence for four additional genes for epileptic encephalopathies. And we haven’t even doubled the cohort size of the initial Epi4K publication yet. Also, for the first time for any neurodevelopmental disorder, we can show that the mutation rate in patients is higher than in the general population. Also, the mutated genes cluster within new functional categories that we suspected, but were not able to pinpoint so far: synaptic function.
Real estate. Here is an interesting technical aspect of the E2 paper that you should be aware of. We used a method that we refer to as real estate analysis to identify genes with more de novo mutations than expected. Basically, this analysis looks at the rate of mutations in each gene taking into account the gene size. This might appear trivial at first sight, but it helps us overcome an important problem. Some genes in our genome are gigantic and always accumulate mutations. TTN, coding for titin, is the prime example. How can we tell innocent TTN mutations from causative mutations in FASN, GABBR2 or RYR3 (all four genes have mutations in 2 patients each)? By taking gene size into account. Performing such an analysis, it becomes clear quite quickly that mutations in TTN are not significant, while FASN, GABBR2 or RYR3 are.
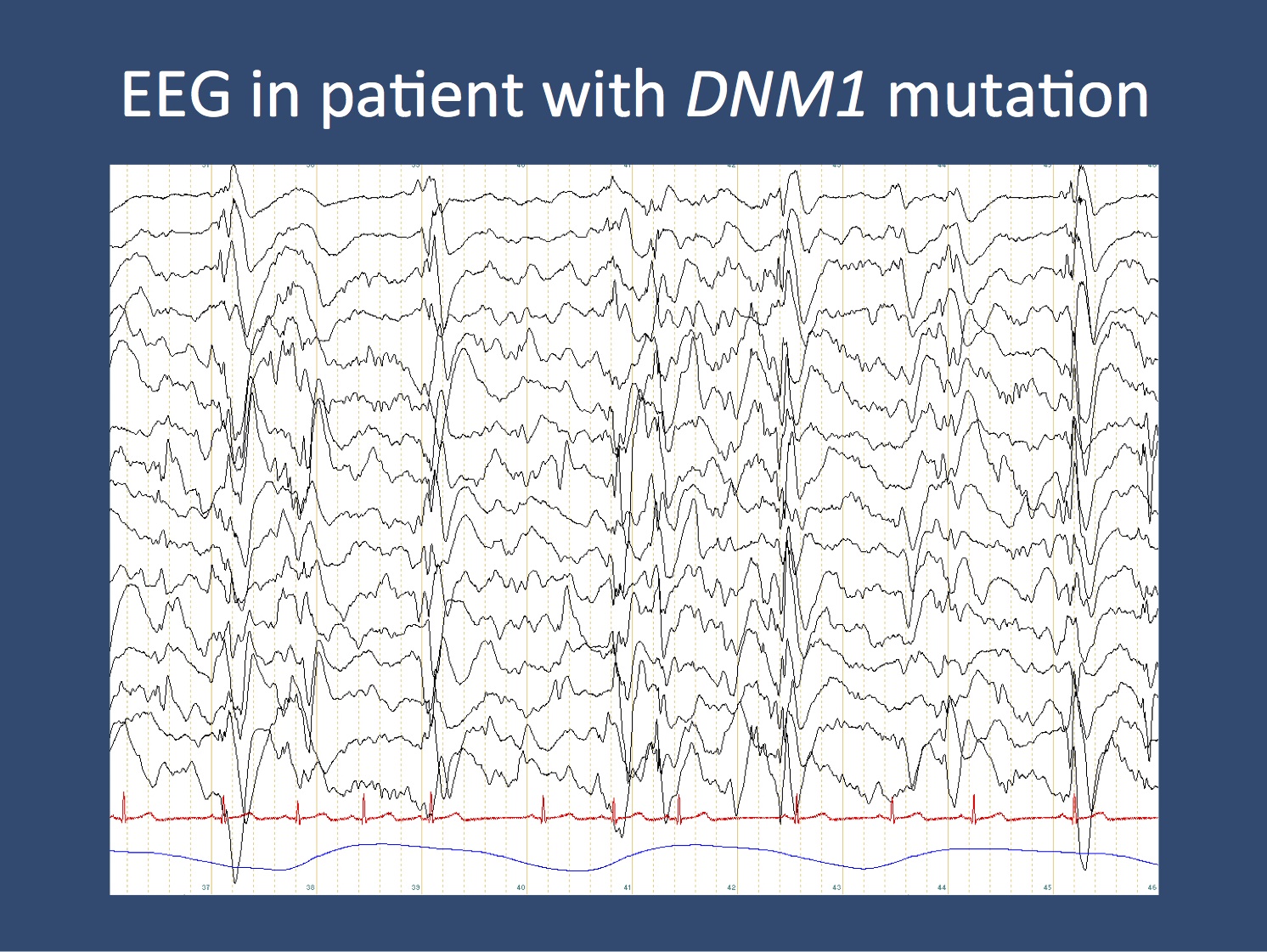
EEG of a patient with a DNM1 mutation, show slow-spike wave during an obtundation status. This EEG is the electrographic hallmark of Lennox-Gastaut Syndrome.
Dynamin 1. The big surprise of the E2 publication is the DNM1 gene coding for Dynamin 1. This small GTPase is involved in severing synaptic vesicles from the plasma membrane (please watch this cartoon on endocytosis.org). Basically, synaptic vesicles constantly merge with the plasma membrane during neuronal firing. In order to recycle vesicles, they are coated with a protein called clathrin. Dynamins then act as the twine that finally severs the recycled vesicle from the synaptic membrane. How this process leads to seizures is still poorly understood. However, we can assume that we are dealing with a process that affects the development of synapses over time. Some patients with DNM1 mutations seem to have intractable epilepsy and a highly active EEG. Figure 2 shows the EEG of a patient with a DNM1 mutation with constant slow-spike wave, the electrographic hallmark of Lennox-Gastaut Syndrome. In total, we identifed 5 patients with de novo DNM1 mutations, accounting for almost 2% of patients with Infantile Spasms and Lennox-Gastaut Syndrome.
The era of gene discovery is over. Let me come back to my provocative opening statement. The era of gene discovery is not over, of course. However, the way we search for genes has changed dramatically. I would assume that we have identified ~10% of all genes for epileptic encephalopathies. However, the remainder of the genes are not unknown, they are simply hidden in the noise. Here is the total list of 429 de novo mutations found in our patients. A large proportion of future epilepsy genes will already be hidden in there. If any of these genes rings a bell to you, please let us know. The E2 study comes with a clear imperative for the research community – COLLABORATE! If we manage to collaborate, genes will find themselves. We simply have to provide the possibility for it. Given the genetic architecture of the epilepsies, future gene findings will not occur in isolation, but through joining datasets and exchanging information. In different words, our current study is only the stepping stone for the next larger analysis of 1000 patient-parent trios.
This is what you need to know. With the currently study, we can securely implicate 26 genes in epileptic encephalopathies. Using a very conservative scoring method, we can explain ~12% of all epileptic encephalopathies. However, a large proportion of mutations in patients with mutations in unique genes would also be called pathogenic by commercial laboratories. The rate of gene discovery in epileptic encephalopathies keeps increasing and we have to think about platforms that would allow for international collaboration on a large scale.
Thank you. This blog post reflects my personal views, but this publication was a joint effort. With this blog post, I would like to say thank you to the EuroEPINOMICS-RES, Epi4K and EPGP community. In particular, on behalf of all coauthors, I would like to thank Sarah Weckhuysen and Erin Heinzen, who coordinated the study and guided us through the submission process. Also, I would like to thank Slave Petrovski, Andrew Allen, and many others at the Epi4K Bionformatics Core at Duke, who where the masterminds behind the analysis. The E2 consortium is currently active in analyzing the published cohort for inherited variants. Stay tuned.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany


Thanks for writing this blog and pointing out the significance of the growing number of genomic studies on epilepsy. You probably saw the recent publications on autism that appeared in Nature. There was a companion publication: A framework for the interpretation of de novo mutation
in human disease, by Samocha etal., that describes the method used for integrated statistical analysis of de novo and other mutation data. Someone should really apply this approach to the extant data available from the E2 consortium.
Hi Doug, apologies for the late reply. I completely agree that this needs to be done, even though I believe that many strategies that aim to put any qualifier on a single mutation in a particular gene that was never identified before eventually use similar strategies. Despite the best prediction tools you might still end up with a level of uncertainty. The measure that we used was a genome-wide approach looking for excess of mutations. This can easily be combined with other methods to interpret mutations.