
Mind the Gap. Ever since its discovery in 2009, SYNGAP1 has been a prominent gene connected to autism and intellectual disability. However, even though probably more than half of all patients with pathogenic SYNGAP1 variants have seizures, it was never a gene that was particularly prominent in the epilepsy field. In a recent publication, we were able to delineate the epilepsy phenotype of patients with pathogenic SYNGAP1 variants, identifying a peculiar combination of generalized seizures types. Here is a blog post about a gene that I admittedly knew very little about before we started working on it.
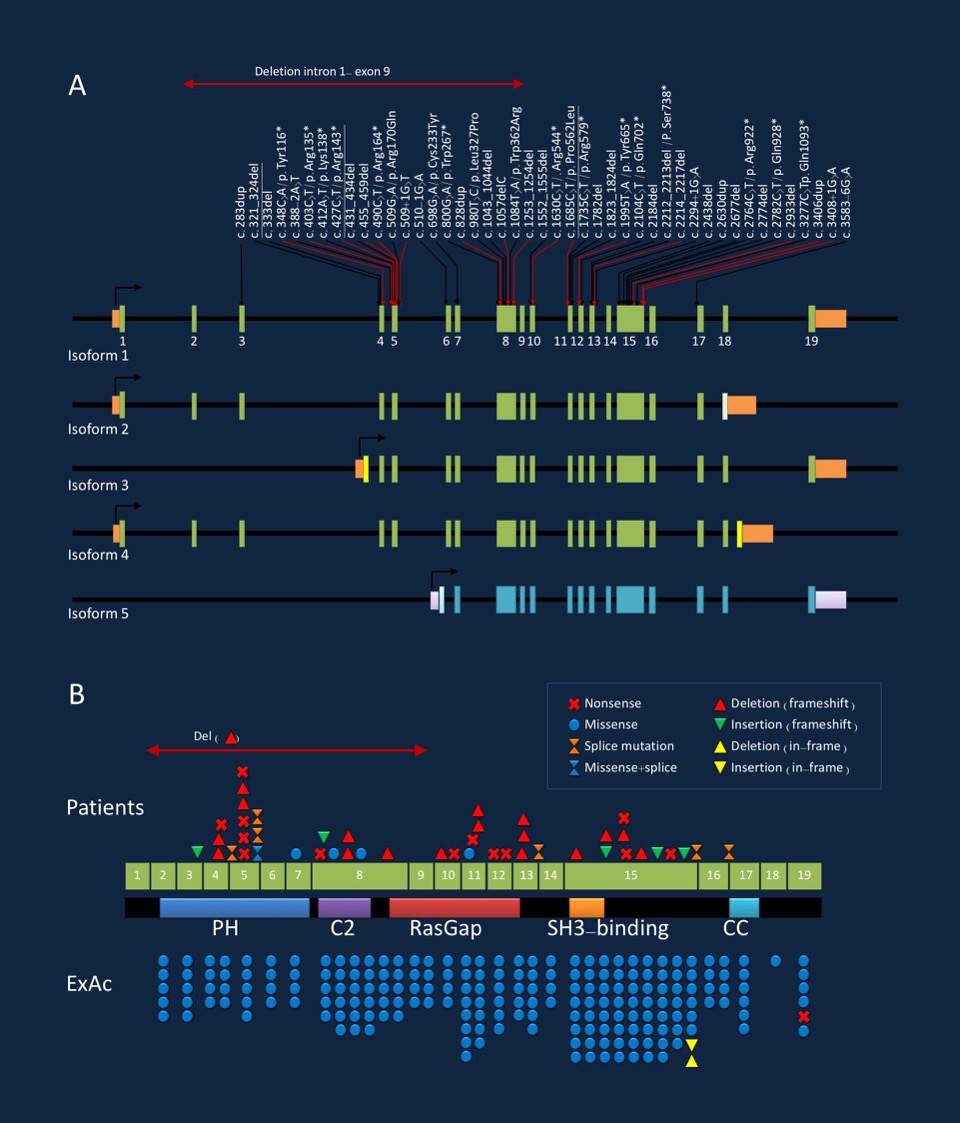
Figure 1. Pathogenic variants in the SYNGAP1 gene with respect to isoforms (A) and protein structure (B). Figure refers to publication by Mignot and collaborators. Figure modified from draft figure provided by the author.
What I learned about SYNGAP1. I was personally surprised by the phenotypic spectrum in patients with SYNGAP1, which revolves around generalized epilepsy with photosensitivity. The study by Mignot and collaborators places SYNGAP1 in a fairly small group of genes that have a phenotype of prominent generalized seizures and the associated EEG features. The other genes in this group would be CHD2, SLC6A1, and – to a certain extent – SCN1A. The SYNGAP1 phenotype appears to be clearly distinct from the spectrum of phenotypes that we usually see with genes for early-onset epileptic encephalopathies, such as CDKL5, SCN2A, SCN8A, or STXBP1.
Epileptic encephalopathy or not. A recent article in Epilepsia cautioned the epilepsy community to use the concept of an epileptic encephalopathy more carefully than we did in the past. The SYNGAP1 phenotype is a good example for this. Basically, the issues raised by Howell and collaborators’ commentary is about the question of whether we consider an epileptic encephalopathy a diagnosis or a mechanism. Many patients with pathogenic variants in SYNGAP1 have intellectual disability and active epilepsy. However, this combination does not automatically generate a diagnosis of an epileptic encephalopathy, as the encephalopathy concept implies that the epileptic activity contributes significantly to the cognitive issues of the patient. As with many complex neurodevelopmental conditions, precisely attributing developmental issues to an underlying epilepsy or an unrelated effect of a pathogenic genetic variant may be difficult.
The SYNGAP1 Epilepsiome page. Visit our SYNGAP1 gene page to learn more about SYNGAP1. There may be some remaining gaps in the SYNGAP1 page that we hope to fill soon. We aim to dynamically add information as it comes up rather than waiting to put information online until a complete review has been completed. The Epilepsiome is meant to be a work in progress.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.

I am so glad to see this post about SYNGAP1 mutations. WE are on the verge of something big and very encouraged by the research that is being spearheaded to make these children’s lives better. I would like to invite all your crew to out First International SYNAGP1 Conference at Texas Children’s Hospital in Houston, Texas USA; November 30 – December 1, 2016. http://www.bridgesyngap.org/#!syngap1-conference/jtqyh
Dear Monica,
thank you so much for letting us know about the activities in the SYNGAP1 community!