
Retinoic receptor. A few years ago, when exome sequencing was still in its infancy, our group in Kiel identified a small de novo in frame deletion in a gene that we didn’t know how to interpret. In frame deletions that do not disturb the reading frame, but simply take one or several amino acids out, are usually less suspicious and are sometimes even filtered out by the algorithms that we and others typically use. We dismissed this finding for several years. However, a year ago, the plot thickened when other groups mentioned that they had found the same gene in their patients, including a family with six affected individuals. In a recent publication, we describe the improbable story of RORB, the latest gene for generalized epilepsies with prominent photosensitivity and absence seizures.
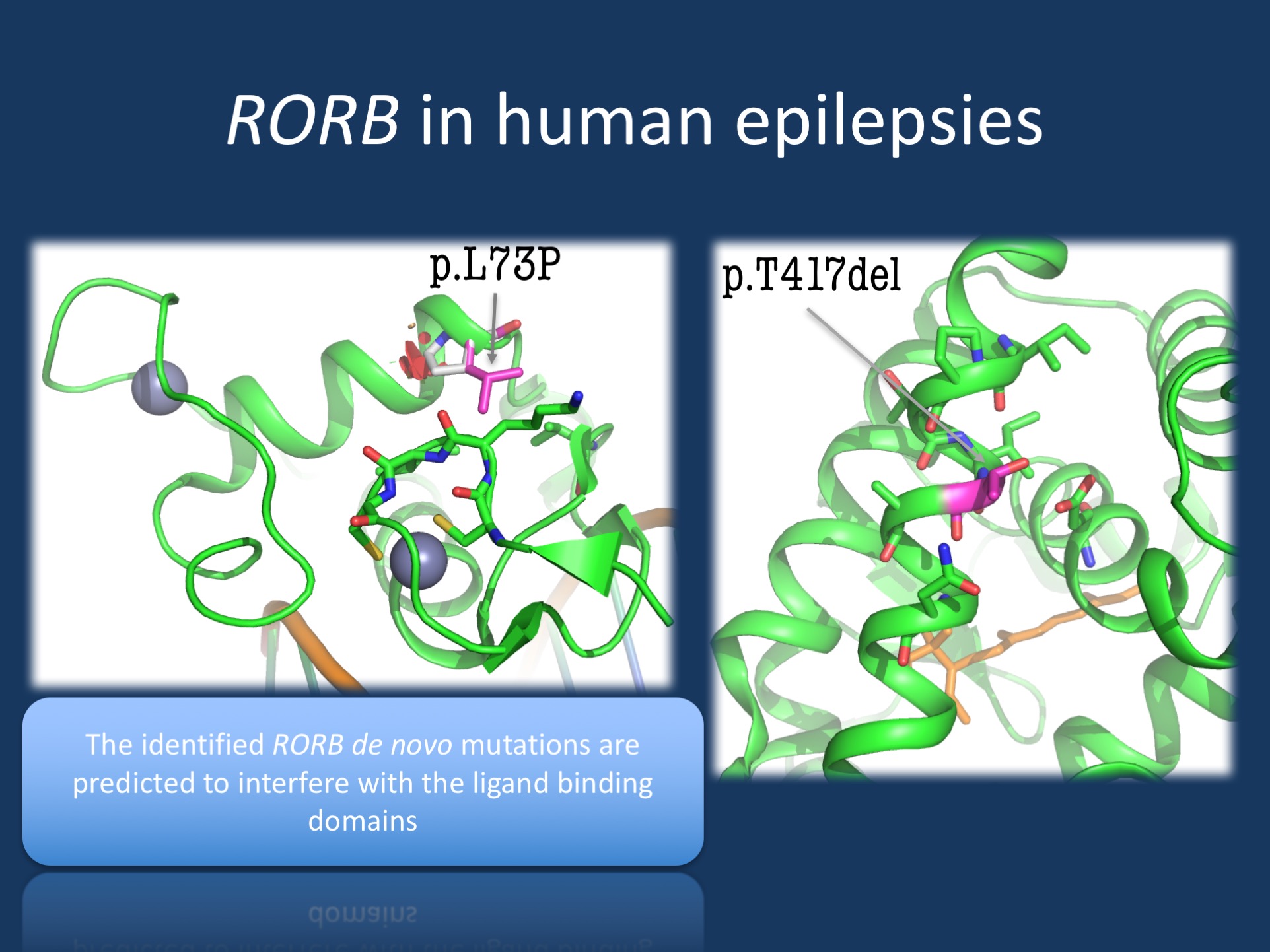
Cartoon structure of the RORB DNA binding domain. Leu73 (de novo mutation) is shown in magenta. The Pro73 (mutation) shown as a white stick with areas of steric clashes as red disks, suggesting that the protein would likely fold much differently in the setting of the L73P mutation. Cartoon structure of RORβ C-terminal ligand binding domain (green) with bound Retinoic Acid. Thr417 (de novo mutation) is show in magenta, suggesting that this position is critically located in the cofactor binding site.
A new class of neurodevelopmental gene. In our recent publication in the European Journal of Human Genetics spearheaded by Gabrielle Rudolf and Pierre Szepetowski, we pull together various pieces of genetic evidence that RORB is implicated in neurodevelopmental disorders, using the current large-scale data sources and collaborative networks that we have. Pulling this type of data together, including data from family studies, de novo mutations identified by exome sequencing, and studies of structural genomic alterations (copy number variations), creates an interesting mosaic of phenotypes and turns genes like RORB into a new type of genes that are supported by a heterogeneous mixture of genetic findings. Ultimately, we leveraged all the data that we had in the community to support the role of this gene in neurodevelopmental disorders. This was ultimately necessary as RORB is a rare gene for these conditions and since it represents an unlikely candidate. So what exactly does this gene do?
Retinoic. RORB is codes for the Retinoid-Related Orphan Receptor-Beta, a nuclear hormone receptor named after its ability to bind retinoic acid in an experimental setting. It is not clear whether retinoic acid is a ligand for this receptor in the human brain and Retinoic acid-related Orphan Receptors (RORs) are considered orphan nuclear receptors that are believed to play a role in various physiological functions, including circadian rhythm. The endogenous ligand for these receptors is unknown. Therefore, in order to implicate this gene in neurodevelopmental disorders, we need sufficient evidence.
Families and sporadics. There are actually not too many epilepsy genes that are found in families with autosomal dominant inheritance and in patients with de novo mutations. Most genes identified in the last five years were implicated in severe epilepsies due to de novo mutations, while gene discovery in familial epilepsies has been lagging behind. The RORB story was somewhat upside down – the current project coalesced around a family with six affected individual with a truncating mutation in RORB that was transmitted in an autosomal dominant fashion. Absence epilepsy and photosensitivity were the predominant phenotypes in this family, setting the stage for the overall phenotypic range that we see in RORB patients. In our current publication we included two more patients de novo mutations in RORB. Also, we included four additional patients with deletions in RORB, including the refined phenotype of a patient we previously reported in a different publication. Back then, we had our first suspicion that there might be more to the gene than we had initially expected.
Phenotypes. The phenotypes of patients with RORB pathogenic variants or RORB deletions is intriguing. Many patients have absence epilepsy and photosensitivity and generalized epilepsy, which somewhat shapes the phenotypic spectrum that we see in patient with RORB. Many patients also have moderate to severe intellectual disability. This combination is somewhat reminiscent of CHD2, a gene that we identified a few years ago. However, other than representing nuclear genes, both genes are involved in different pathways and do not have an immediate connection.
Haploinsufficiency. Coming back to our patient with the in frame deletion in RORB, how can we reconcile the seemingly benign de novo variant with the profound effect on RORB function? We modelled the molecular structure of RORB and found that the Thr417 protein residue is very important for the tertiary structure of the protein. Losing this amino acid residue is predicted to cause a significant structural change in a binding site for cofactors, likely interfering with RORB function in a profound way. Taken together, even though the deletion appeared benign at first, it was predicted to result in a profound structural change of the RORB protein, a change that may be as severe as a deletion or truncating mutation in the RORB protein.
What you need to know. Combining data from family studies, de novo alterations, and microdeletions, we provide evidence for a role of RORB in neurodevelopmental disorders, particularly generalized epilepsies with photosensitivity. RORB is a protein of unknown function, but haploinsufficiency of the RORB gene appears to be the pathogenic mechanism. RORB encephalopathy appears to be very rare, but given our ability to pull together data on this gene in a timeframe of less than a year, we would expect patients to be identified in the next few years, which will allow us to better assess the phenotypic spectrum.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
