
Ketogenic. Several patients with Myoclonic Astatic Epilepsy (MAE) have a surprisingly positive response to the ketogenic diet, even after failing several antiepileptic medications. Given this observation, it is obvious to assume that SCL2A1 mutations play a significant role in MAE, and some earlier studies seemed to suggest that up to 5% of MAE patients carry SLC2A1 mutations. However, in a recent study, we failed to demonstrate a connection between SLC2A1 mutations and Myoclonic Astatic Epilepsy (MAE) in a large cohort of patients. Read more on why the genetics of MAE is an ongoing mystery and why we need a new approach to “keto-genetics”.
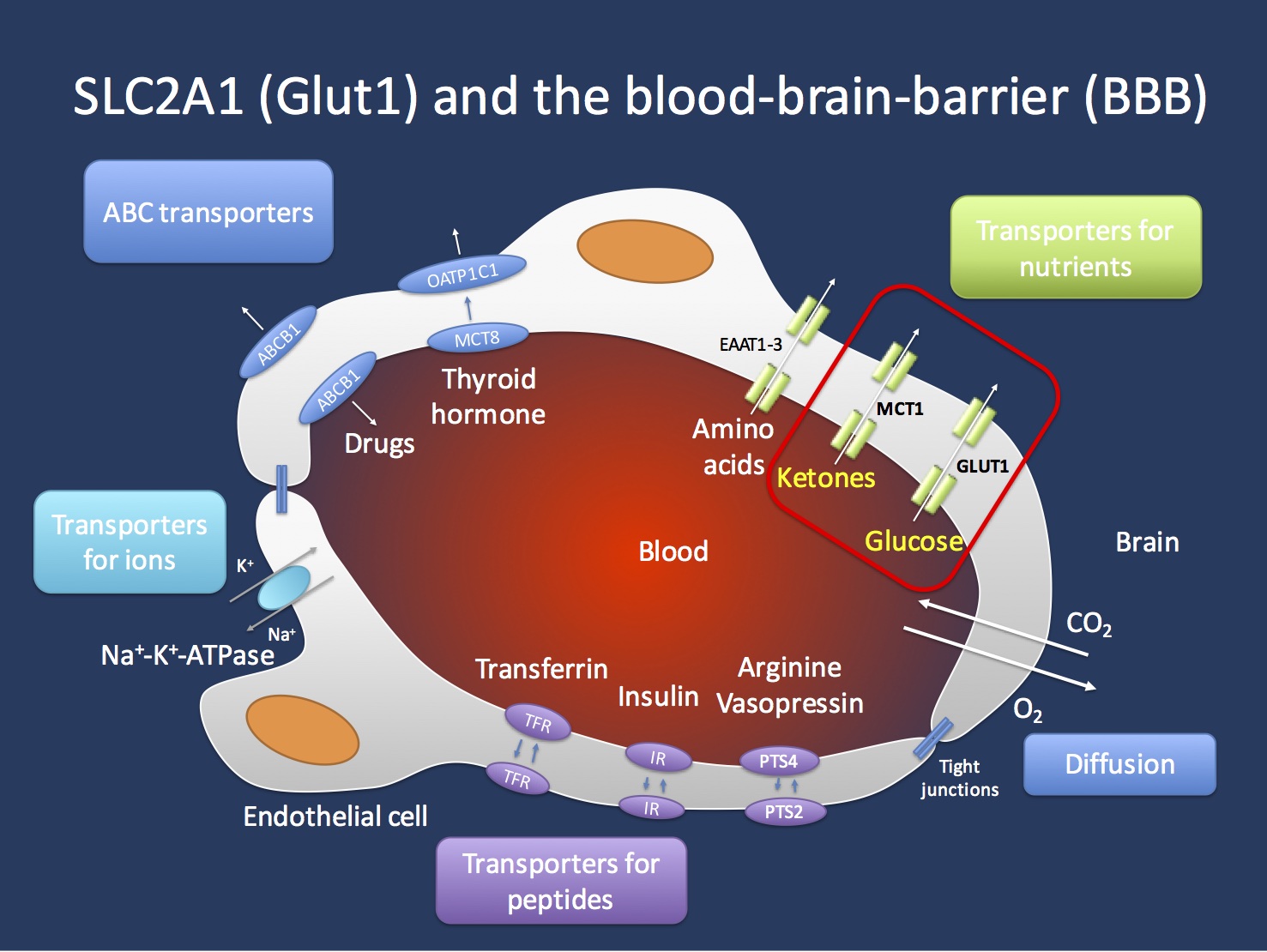
Transport across the blood brain barrier (BBB). The endothelial cells in the Central Nervous System are important gatekeepers of transport into the CNS. The GLUT1 transporter encoded by the SCL2A1 gene is the main transporter for glucose and mutations in the SLC2A1 gene lead to GLUT1 Deficiency Syndrome. This condition can be treated with the ketogenic diet that allows the brain to primarily use ketones as an alternate source of energy. Ketones are primarily transported through the MCT1 transporter, which is encoded by the SLC16A1 gene. Figure inspired by Zlokovic, Nature Reviews Neuroscience 12, 723-738 (December 2011).
Chasing the Doose gene. Myoclonic Astatic Epilepsy is a perplexing epilepsy syndrome that typically presents in preschool children with a fulminant onset. Children often present with atonic seizures (“drop attacks”) and they often have additional seizure types including absence seizures, myoclonic seizures, and generalized tonic-clonic seizures. The epilepsy and especially the epileptic activity on EEG is frequently severe enough to result in developmental problems or even regression, a phenomenon referred to as epileptic encephalopathy. Myoclonic Astatic Epilepsy (MAE) or Doose Syndrome is a generalized epilepsy and neuroimaging (MRI) is almost always unremarkable. MAE is strongly suspected to be a genetic epilepsy. However, the genetic cause in most patients is unknown.
Known MAE genes. There is some evidence for a minor role of relatively common epilepsy genes such as SCN1A or the epilepsy-related microdeletions. Earlier this year, we described mutations in SLC6A1 in patients with Doose Syndrome, but mutation in this gene also only explain a small fraction of patients and was only found in patients with significant developmental delays prior to epilepsy onset. Other genes implicated in single patients with MAE are SCN2A, STX1B, GABRG2, and CHD2. Mutations in SCL2A1 had been previously described in up to 5% of patients in some case series. However, most patients with SCL2A1 mutations had additional prominent features such as movement disorders. The RES consortium is continuing our efforts to perform exome sequencing in a large cohort of patients with MAE and collect data from patients who have already been sequenced.
Lack of SLC2A1 mutations. In the study by Larsen and collaborators, we examined 120 patients with MAE, including the entire RES MAE cohort. In the 120 patients examined, we did not find a single patient with an SLC2A1 mutation, suggesting that the frequency of GLUT1 deficiency in MAE without additional features is very low. Motivated by the prior reports on SCL2A1 mutations in MAE, there has been a heightened awareness in the field to think of GLUT1 in patients with MAE and atypical generalized epilepsies. The negative findings in our study should not be taken as a justification to let our guards down – in fact, Glut1 deficiency remains one of the top differential diagnosis in patients with refractory or early-onset generalized epilepsies. In our study, we identified six SLC2A1 mutations in other types of epilepsies, including early-onset absence epilepsies. Given that the ketogenic diet is an available causative treatment option, even a low prior probability will justify testing. In summary, in atypical, refractory, or early-onset generalized epilepsies, think Glut1.
Keto-genetics. The Myoclonic Astatic Epilepsies are among a group of epilepsies that often respond very favorably to the ketogenic diet. While it is obvious to assume that these responders may have hidden Glut1 deficiencies, this is often not the case, and the group of MAE patients responding remarkably well to the ketogenic diet extends beyond the very small fraction of patients with Glut1 deficiencies. Admittedly, we did not collect information on how many MAE patients had a good response to the ketogenic diet in our study. Also, to my knowledge, there is not study that systematically examined the frequency of Glut1 deficiency in patients responding well to the ketogenic diet. Having said this, you may wonder whether it is time for a fresh approach to “keto-genetics”, examining possible genetic determinants of response to the ketogenic diet. The group of patients responding to the ketogenic diet spans across various genetic epilepsies, including patients with Dravet Syndrome and mutations in SCN1A or other genetic epileptic encephalopathies. Maybe it is reasonable to ask whether some of the response or non-response may in part be affected by modifying genetic factors.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.

our daughter has a mutated KIAA2022 gene, and she has Doose. There is no mention of this gene mutation in your article.
http://epilepsygenetics.net/2016/07/13/where-the-genes-have-no-names-kiaa2022-in-epileptic-encephalopathy/