
STXBP1. The next candidate in our weekly review of epilepsy genes is STXBP1. This gene was initially discovered in 2008 as cause for Ohtahara Syndrome. Ever since, it has become one of the most prominent genes for epileptic encephalopathies so far. Here is what you need to know about STXBP1 in 2015.
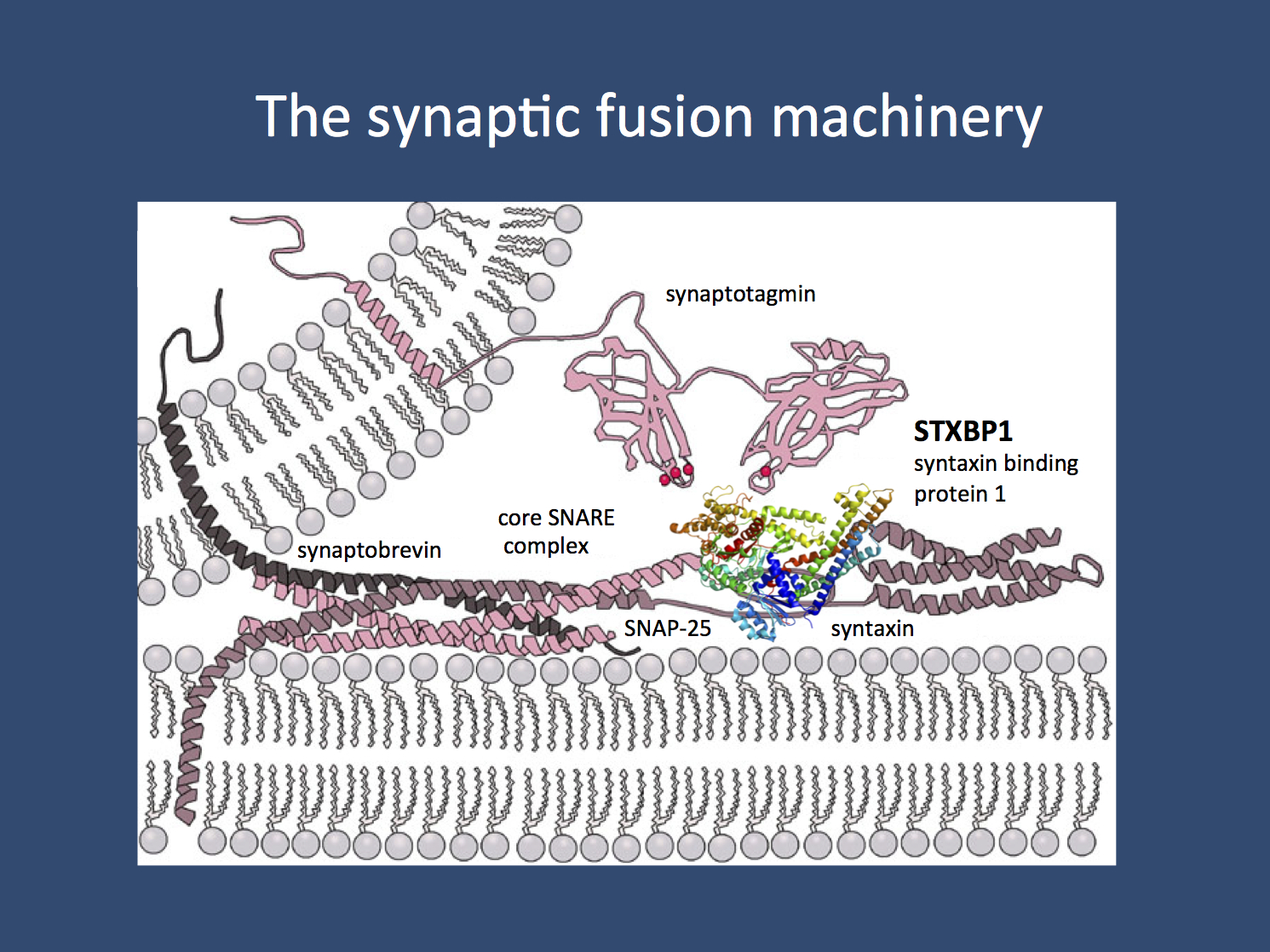
Vesicle fusion. The SNAREpins enable the final act of vesicle fusion that overcomes the repulsion of two lipid bilayers. STXBP1 is one of the proteins interaction with syntaxin, one of three proteins thought to be involved in this process. (Images are from Wikipedia under the GNU Free Documentation License)
Phenotype. STXBP1 encephalopathy was initially discovered in patients with Ohtahara Syndrome and it was initially felt that the phenotype would be associated exclusively with a severe neonatal epilepsy. Since the initial description by Saitsu and collaborators, the phenotype has significantly broadened. In 2015, STXBP1 encephalopathy has emerged as one of the most common genetic causes of epileptic encephalopathy with phenotypes ranging from severe neonatal epilepsy to infantile-onset epilepsy such as Dravet Syndrome. Some publications have pointed out that the epilepsy in STXBP1 encephalopathy can be quite dynamic, starting very abruptly with a highly pathological EEG pattern, sometimes with a dramatic improvement after the age of 6 months that may not necessarily be due to the antiepileptic medication. Additionally, many patients with STXBP1 mutations have been described with non-epileptic movement disorders, including truncal and limb ataxia, generalized tremors, and dystonia. Interestingly, STXBP1 may not exclusively be associated with epilepsy phenotypes, as some patients with intellectual disability without seizures have also been reported.
Genotype. STXBP1 encephalopathy is due to haploinsufficiency of the gene for the syntaxin-binding protein 1. Various truncating mutations and deletions have been identified in this protein and from what we know so far, there is no consistent genotype/phenotype correlation even though larger studies have not been published to date. There are some recurrent mutations in the STXBP1 gene listed in ClinVar, but the importance of this is not known today.
Mechanism. Syntaxin-binding protein 1 is a part of the synaptic fusion machinery that enables vesicles to fuse with the plasma membrane. In a previous post, we have used the analogy of trying to fuse soap bubbles in a bubble bath. Given the hydrophilic and hydrophobic aspects of lipid bilayers, fusion of bubbles always required energy. In cells, the so-called minimal fusion machinery provide the final “push” for the vesicle to fuse with the membrane through protein that twist around each other and pull the vesicle close enough to fuse.
Here are the five things about STXBP1 that you need to know in 2015:
Dravet Syndrome. When STXBP1 first entered the scene, it was believed to be a gene for Ohtahara syndrome, a severe, early-onset epilepsy. However, over time, other phenotypes have emerged, expanding the phenotype to other epilepsies. One of the more unexpected phenotypes was SCN1A-negative Dravet Syndrome that we described in the publication of Carvill and collaborators in 2014. There are few genes with such a broad phenotypic spectrum.
Keppra. In a recent publication in Pediatric Neurology, Dilena and collaborators report on a dramatic effect on the seizure burden in a one months old child with STXBP1-related Ohtahara Syndrome and they suggested a causal mechanism and recommended Keppra as a first-line medication for this condition. Having seen several children with STXBP1-related epilepsy fail Keppra, I was surprised about the strong wording in this article. However, it shows that the epilepsy in STXBP1 encephalopathy is dynamic and can respond to treatment in a surprising and possibly non-specific fashion in some children.
Movement disorders. The STXBP1 phenotypic spectrum has been found to extend beyond epilepsy and developmental issues. Various movement disorders have been reported that occur as early as the neonatal period and in a publication by Keogh and collaborators, two patients were described with Parkinsonism. These reports emphasize that STXBP1 plays an important role in subcortical brain structures, sometimes resulting in a complex phenotype that encompasses epilepsy and non-epileptic features.
Mechanism. Syntaxin-binding protein 1 is also known as Munc18-1 and this protein has been a late discovery in the protein affiliated with fusion of synaptic vesicles. Initially, only the three main components of the fusion machinery were thought to participate in fusion: syntaxin, synaptobrevin, and SNAP25. A 2013 Science paper by Ma and collaborators, however, has placed Munc18-1 very prominently in the early process of neurotransmitter release. Basically, by binding to syntaxin, Munc18-1 orchestrates the assembly of the other components.
Related genes. Ever since the initial discovery of STXBP1 in Ohtahara Syndrome, several other genes coding for proteins in the presynaptic fusion machinery have been identified as genes for human epilepsies. Most prominently, these include PRRT2, the most frequent genetic cause of benign infantile seizures. Also, mutations in STX1B and SNAP25 have been found to be mutated in patients with genetic epilepsies. This emerging picture suggests that disruption of the regular function of presynaptic proteins may results in epilepsy. This mechanism may be counterintuitive at first glance given that global impairment of neurotransmitter release should primarily affect the excitatory neurons. However, it is the current working hypothesis that GABAergic interneurons may be more vulnerable, resulting in overall reduced inhibition.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
