
BPAN. We have selected WDR45 to be our Epilepsiome gene of the week. WDR45 was initially identified as the causative gene for a rare phenotype referred to as static encephalopathy with neurodegeneration in adulthood (SENDA), which belongs to a group of neurodegenerative disorders that have accumulation of iron in the CNS as the common feature. In contrast to the narrow and very specific phenotype in most other disorders in this group, the phenotypic spectrum of WDR45 has expanded significantly since the initial discovery in 2013. Mutation in WDR45 can be identified in patients with a broad range of neurodevelopmental phenotypes including epileptic encephalopathies.
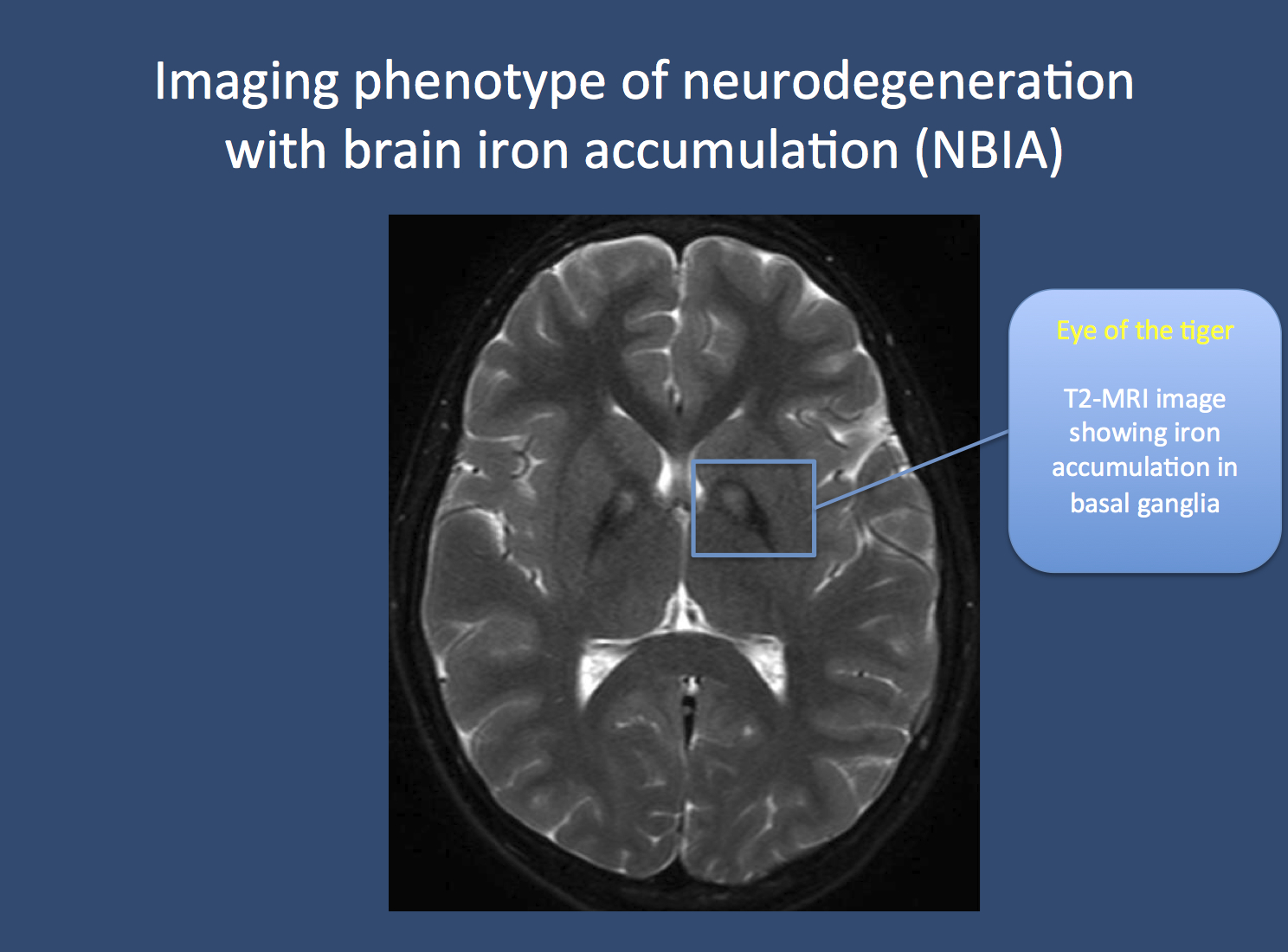
Figure. The eye of the tiger in NBIA in a patient with pantothenate kinase-associated neurodegeneration (PKAN). Neuroimaging in patients with PKAN and other NBIA is usually associated with a pathognomonic finding. Due to the accumulation of iron in the basal ganglia, two black spots can be seen, which is referred to as the “eye of the tiger” sign. PKAN is part of a group of disorders referred to as neurodegeneration with brain iron accumulation. Image from Wikimedia commons (http://commons.wikimedia.org/wiki/File:Pkan-basal-ganglia-MRI.JPG), within the framework of the Creative Commons license.
NBIA. We last wrote about WDR45 in 2013 when a publication by Hayflick and collaborators introduced the concept of beta-propeller associated neurodegeneration (BPAN) for the various phenotypes caused by WDR45 mutations. I have adapted our initial blog post to add some of the most relevant updates in 2015, particularly the emerging male WDR45 phenotype. In 2013, Hayflick and collaborators suggested that, in childhood, the phenotype shares similarities with atypical Rett syndrome and other epileptic encephalopathies, which raised the general question whether some of the epileptic encephalopathies presenting in childhood may be the initial presenting feature of neurodegenerative disorders. BPAN belongs to a group of conditions referred to as neurodegeneration with brain iron accumulation (NBIA), a group of mainly recessive disorders that present with progressive dystonia and dementia. The common feature of these diseases is the excessive accumulation of iron in the basal ganglia. NBIA is very rare and usually not discussed in the context of epilepsy. PKAN – pantothenate kinase-associated neurodegeneration is historically considered the most prominent NBIA subtype as it was the first NBIA to be identified.
PKAN, INAD, MPAN, BPAN. In 1922, Hallervorden and Spatz described a familial disease that was characterized by a severe and progressive neurodegenerative disease starting between the ages of 7 and 15. Patients were found to have progressive dementia, spasticity, rigidity, dystonia, and choreoathetosis. The disease was later found to be accompanied by excessive deposits of iron in the basal ganglia. These deposits appeared as dark spots on the MRI, which became known as the “eye of the tiger” sign. A mutation in PANK2 coding for pantothenate kinase was found to cause this disease, which was subsequently renamed PKAN – pantothenate kinase-associated neurodegeneration. The name Hallervorden-Spatz disease is no longer preferred, given Hallervorden and Spatz’s complicity in murderous Nazi programs. Over time, other diseases with related imaging findings were identified, including infantile neuroaxonal dystrophy (INAD) due to mutations in PLA2G6 and mitochondrial membrane protein-associated neurodegeneration (MPAN) with mutations in C19orf12. Both disorders are characterized by progressive dementia and movement disorders, but may present earlier than PKAN. In 2013, WDR45 mutations were found to underlie a novel NBIA subtype, which was initially termed static encephalopathy with neurodegeneration in adulthood (SENDA). “Static encephalopathy” referred to the first phase of the disease, which resembled various neurological disorders in children, including epileptic encephalopathies.
From SENDA to BPAN. In their 2013 publication, Hayflick and colleagues reviewed the clinical features of 23 patients with mutations in WDR45 and described the phenotypes in detail. WDR45 is a so-called beta-propeller protein which is assumed to be involved in autophagy, the breakdown of cellular components through lysosomes. Given the properties of the protein involved, Hayflick and collagues opted to call the phenotype BPAN, beta-propeller associated neurodegeneration, as “static encephalopathy” did not entirely capture the early presentation of the disease. In the early phase of the disease, patients were found to have global developmental delay, epilepsy and stereotypies, i.e. repetitive movements such as hand flapping. Particularly the latter reminded the authors of other diseases including Rett Syndrome and atypical Rett Syndrome. The epilepsy phenotypes comprised myoclonic, astatic and absence seizures. Furthermore, the authors suggest that neuroimaging might be normal in childhood. In young adulthood, dystonia, parkinsonism and progressive dementia was noted and the MRI showed basal ganglia iron deposits. WDR45 is located on the X-chromosome and all patients had de novo mutations in this gene, turning BPAN into the only dominant form of NBIA.
WDR45 – a 2015 update. Even though the 2013 publication by Hayflick and colleagues already hinted at a strong phenotypic overlap of WDR45 encephalopathy and the epileptic encephalopathies, this concept became more widely known with the publication of several case reports, such as a case report in a patient with early-onset epileptic encephalopathy that we discussed earlier this year. As WDR45 is located on the X chromosome, it is assumed that males are typically affected more severely and may have a neonatal or early infantile presentation of profound delays, seizures, and neuroimaging abnormalities. Affected females may present with developmental delays and the static encephalopathy that initially gave rise to the concept of SENDA.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
