
PME. The progressive myoclonus epilepsies (PME) are a particular subtype of seizure disorders characterized by progressive myoclonus, generalized seizures and cognitive deterioration. Known causes of PME include recessive mutations in several well-known genes, but the genetic cause is unknown in a significant proportion of patients. Now, in a recent paper in Nature Genetics, de novo mutations in KCNC1 are identified as a novel cause of progressive myoclonus epilepsies. In addition to elucidating the genetic basis in a significant subset of patients with PME, the authors demonstrate that de novo mutations play an important role in a group of diseases usually thought to be recessive.
The PMEs. The progressive myoclonus epilepsies are a group of seizure disorders that initially present with therapy-resistant myoclonus that gets worse over time and can be accompanied by generalized tonic-clonic seizures and cognitive decline. The PMEs are usually thought of as a distinct group of epilepsies with a genetic basis and is usually divided into two larger groups. Unverricht-Lundborg disease (ULD) usually has preserved cognition until late into the disease, while disorders such as Lafora body disease show prominent neurological decline and dementia. Only a few years ago, the genetics of ULD-type PME was simple, as an expansion of a dodecamer repeat in the 5’-UTR of the Cystatin B (CSTB) gene was the only known genetic cause. However, with the discovery of SCARB2, PRICKLE1 and GOSR2, additional genes were found in ULD-type PMEs. The Lafora-type PMEs were traditionally thought of as more heterogeneous as several storage disorders including sialidosis, mitochondrial disorders or neuronal ceroid lipofuscinosis may present as progressive myoclonus epilepsies. Muona and collaborators examined a cohort of 84 unsolved cases of PME using exome sequencing.
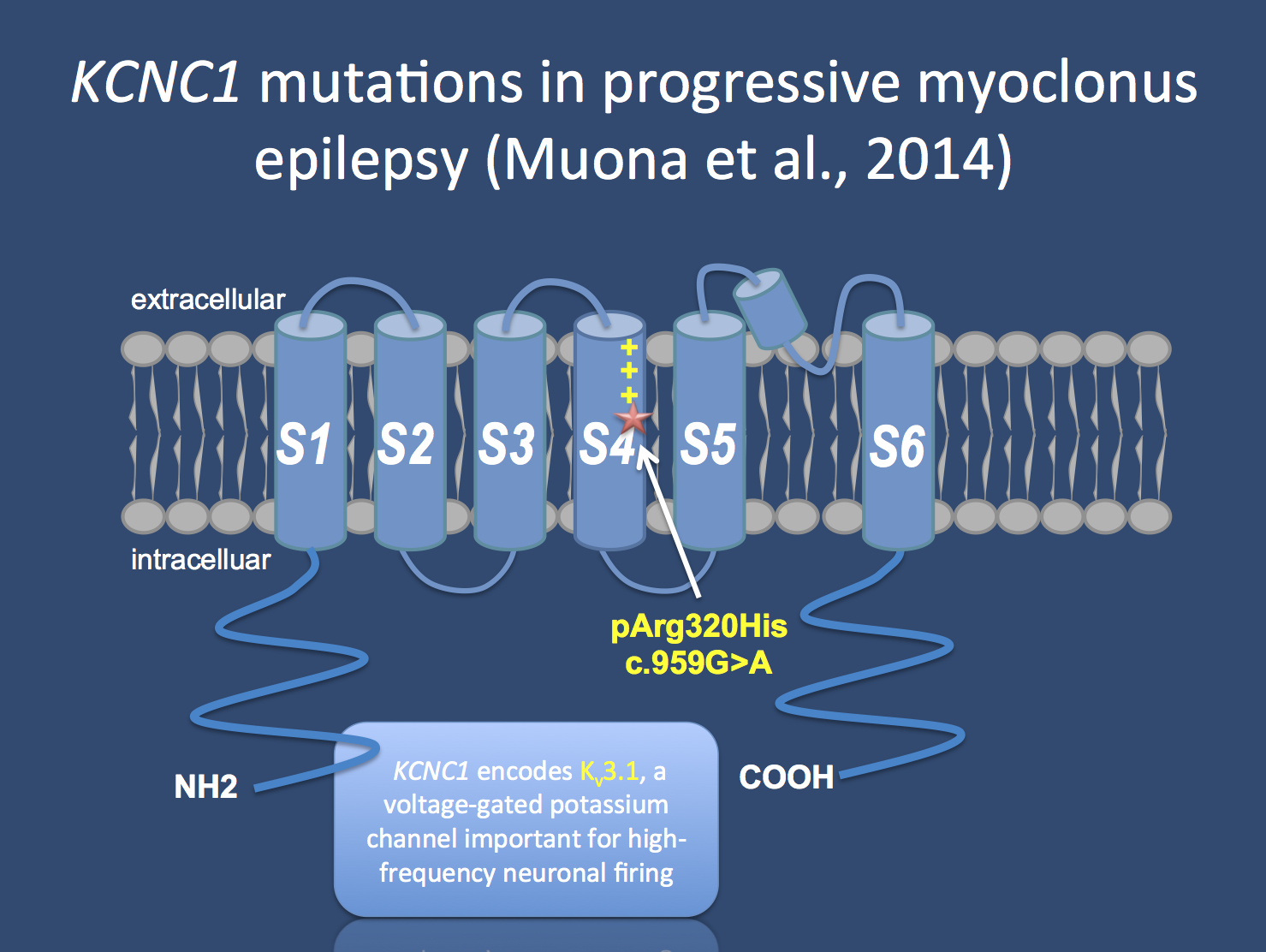
The KCNC1 channel is a brain-expressed voltage gated potassium channel working as a “delayed rectifier”. De novo mutations in KCNC1 are a major cause of progressive myoclonus epilepsies and a single recurrent mutation is found in patients, which acts in a dominant negative fashion.
Typical versus atypical. At first, the authors looked at known PME genes or possible candidate genes and found pathogenic or probably pathogenic mutations in 15/84 patients, which is almost 20%. This cohort was previously screened for CSTB mutations (ULD) and mutations in SCARB2 and GOSR2 in a significant proportion of patients. The authors identified known disease genes in 10 patients including NHLRC1 and EPM2A (Lafora disease), NEU1 (sialidosis), AFG3L2 (spinocerebellar ataxia with PME), CLN6 (adult form of NCL), and SERPINI1 (PME with neuroserpin inclusion bodies). However, the remaining 5 patients with an identified molecular cause were very interesting. The authors identified one family with a recessive mutation in TBC1D24, a gene with an expanding phenotypic spectrum. PME, however, had not been described as a phenotype. Also, they identified two families with compound heterozygous mutations in SACS, the gene for the autosomal recessive spastic ataxia Charlevoix Saguenay. PME was also not previously described as a phenotype of this ataxia syndrome that may mimic cerebral palsy. Furthermore, the authors identified a mutation in the PRNP gene, a known cause of Gerstmann-Straussler-Schenker disease, a human prion disease. Finally a single de novo mutation in SCN1A was found in a patient initially classified as PME. However, upon detailed review of the phenotype, this patient was found to have features of Dravet Syndrome. While the screen for known disease genes explained almost 20% of cases, the real surprise of the study by Muona and collaborators was a recurrent de novo mutation.
KCNC1 and neuronal firing. Muona and collaborators identified a de novo mutation in KCNC1 in 13 patients with PME, identifying a major novel cause of the ULD-type PMEs. KCNC1 is a brain-expression potassium channel that works as a ‘delayed rectifier’ and is a member of the Kv3 potassium channel family. This family of potassium channels has specific electrophysiological properties that are important for high-frequency neuronal firing. While it is difficult to estimate the overall contribution of KCNC1 to the overall group of PMEs given that the cohorts was previously screened for CTSB, the proportion explained by KCNC1 almost approaches the cumulative proportion of all other genetic causes, suggesting that this is a major new disease gene. Also, all patients have a single recurrent de novo mutation (p.Arg320His) that affects the voltage sensor of this channel and acts in a dominant negative way. Basically, while the mutation is only on one allele, it almost acts as a knockout, as the mutation drags down the function of the remaining allele. The authors show that the function of the channel is actually reduced by 80% when the mutant channel is present in a 1:1 ratio with wildtype KCNC1.
This is what you need to know. KCNC1 mutations are a novel, major cause of the progressive myoclonus epilepsies. All patients have a single, recurrent de novo mutation. While the genetic basis of PME was previously thought to be recessive, the study by Muona and collaborators emphasizes an emerging role for de novo mutations in this group of diseases. This is quite remarkable as some PMEs have a relatively late onset and as de novo mutations were thought to be the domain of early-onset epilepsies. The current study raises the issue whether it is worthwhile examining the de novo paradigm also for a broader group of epilepsies.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany

