
The return of TBC1D24. In 2010, the TBC1D24 gene was the first gene for human epilepsies to be discovered through next generation sequencing techniques. Ever since, this gene has been a mystery, as the phenotypes of the families with recessive mutations in this gene varied widely. Now, a recent paper in Lancet Neurology finds recessive TBC1D24 mutations in a large proportion of patients with DOORS syndrome, a rare distinct autosomal recessive syndrome with deafness, onychodystrophy, osteodystrophy, intellectual disability (mental retardation), and seizures. This finding demonstrates that we have only just scratched the surface of the complicated genetic architecture of human epilepsies.
TBC1D24. The TBC1D24 protein is expressed in the Central Nervous System and is named for its TBC domain. This conserved part of a protein is usually found when a protein interacts with a GTPase, which suggests a function in cellular signaling with respect to growth or vesicle regulation. For TBC1D24, an interaction with ARF6, a small GTPase involved in neuronal cell polarity and regulation of exocytosis and endocytosis, has been described. This is as much as we know about the TBC1D24 protein to date.
TBC1D24 and epilepsy. The TBC1D24 gene was first found to be involved in human epilepsy in 2010 when two simultaneous publications implicated this gene in two different epilepsy syndromes. In a family from Italy, mutations were identified in a relatively benign Familial Infantile Myoclonic Epilepsy (FIME). In parallel, mutations were found in a consanguineous Arab-Israeli family with focal epilepsy, intellectual disability, and cortical thickening. Recessive mutations or compound heterozygous mutations in the same gene were subsequently identified in a family with severe epileptic encephalopathy with neurodegeneration and in a family with familial malignant migration partial seizures of infancy (MMPSI). These findings were already puzzling with respect to the phenotypic variability seen in families with mutations in this gene. Now, Campeau and collaborators identify mutations in TBC1D24 in DOORS syndrome.
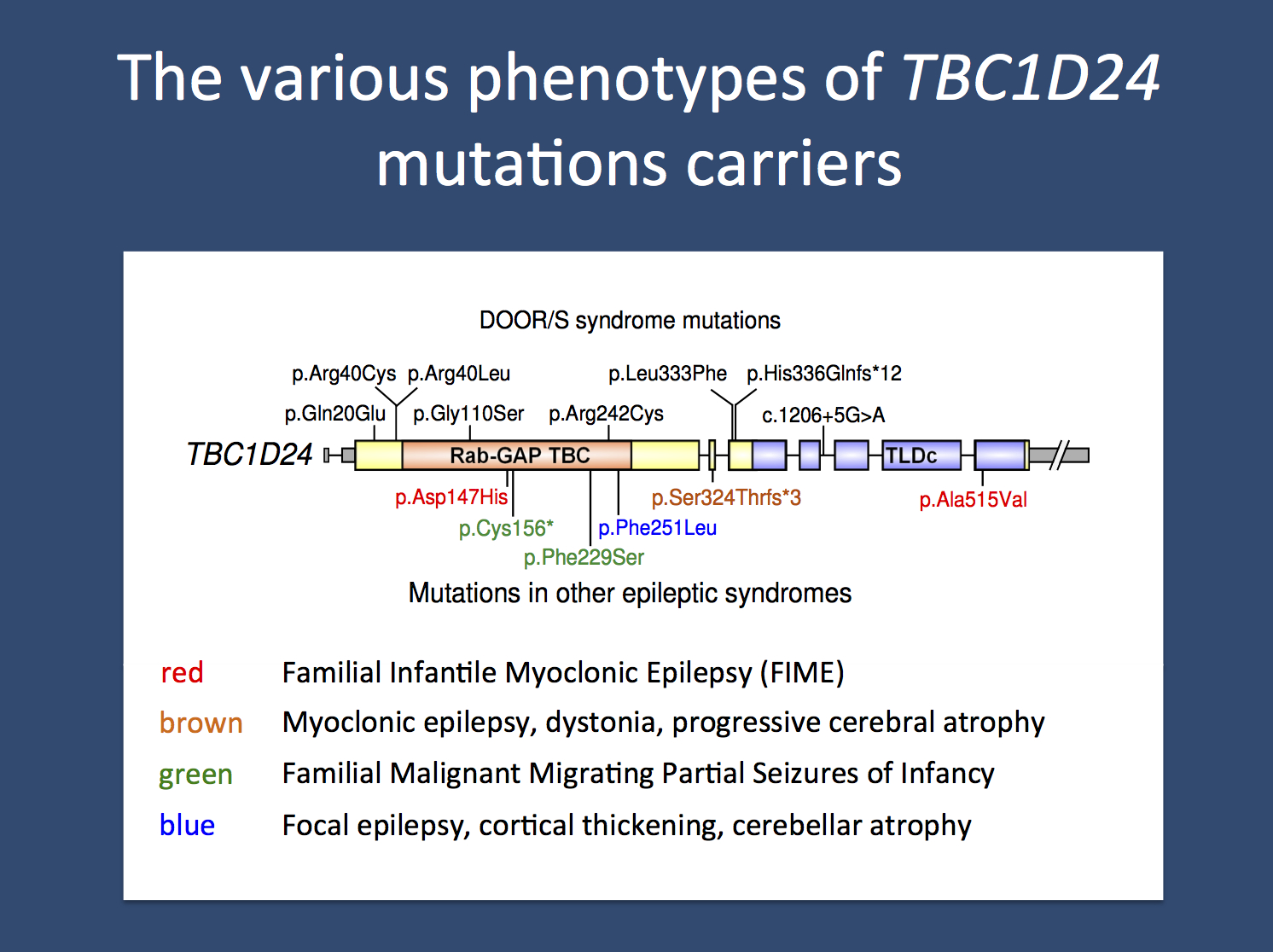
The various recessive and compound heterozygous epilepsy phenotypes due to mutations in TBC1D24.
DOORS and TBC1D24. The acronym DOORS refers to the combination of deafness, onychodystrophy, osteodystrophy, intellectual disability (mental retardation), and seizures. DOORS syndrome is a rare, autosomal recessive syndrome that has epilepsy as an inconsistent feature. Accordingly, the “S” in DOORS is sometimes omitted (DOOR). Onychodystrophy refers to malformation of the nails, which can be malformed or even absent. Osteodystrophy refers to malformation of the skeletal systems. In patients with DOORS Syndrome, usually the hands and feet are affected. In one third of patients, a triphalangeal thumb is observed, a thumb with three instead of the usual two joints. The epilepsy in DOORS syndrome usually starts in the first year of life and can be refractory to treatment. Using exome sequencing in 26 families with DOORS Syndrome, Campeau and collaborators identified mutations in TBC1D24 in 9 families. These findings suggest that the role of TBC1D24 extends beyond the Central Nervous System and that it is also involved in the formation of bone and skin structures. With respect to the heterogenous phenotypes, TBC1D24 is the first example of a recessive gene with a broad phenotypic range, a novelty for epilepsy-related gene findings.
Homogeneous phenotypes in recessive epilepsies. Traditionally, recessive epilepsy syndromes are rare in contrast to the genetic epilepsies caused by de novo and dominant mutations. We usually associate recessive epilepsy syndromes with severe, progressive epilepsy, such as the progressive myoclonus epilepsies or epileptic encephalopathies due to neurometabolic disorders, mitochondrial disorders, storage disorders, or congenital disorders of glycosylation (CDG). As discussed in previous posts, with the exception of the widening spectrum of congenital disorders of glycosylation, these conditions are usually quite distinct and do not have the broad phenotypic range as seen in dominant epilepsies. In summary, the “hidden neurometabolic” hypothesis has found little support so far with the exception of rare, unscreened consanguineous families. Therefore, the identification of de novo mutations has been the focus of the field over the last two years. TBC1D24 makes us reconsider this choice.
Recessive and pleiotropy. The example of TBC1D24 suggests that certain recessive genes may also present with a very broad phenotypic range. Possibly, TBC1D24 is only one example of an entire class of genes with similar properties that may span various epilepsy phenotypes and even various neurodevelopmental disorders. As with TBC1D24, mutations in these genes are likely to be rare, otherwise they would have already been identified. Nevertheless, this observation suggests that a pooled analysis of various epilepsy syndromes and possibly other neurodevelopmental phenotypes with respect to existing trio exome data might be worthwhile. We might in fact discover additional genes with similar properties as TBC1D24, genes that we didn’t really think would exist.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany


Pingback: Microcephaly, WDR62, and how to analyze recessive epilepsy families | Beyond the Ion Channel
Pingback: SLC25A22, migrating seizures and mitochrondial glutamate deficiency | Beyond the Ion Channel
Pingback: The OMIM epileptic encephalopathy genes – a 2014 review | Beyond the Ion Channel