
Synaptic transmission. Over the last several years, pathogenic variants in multiple genes involved in synaptic transmission have been identified in early-onset epilepsies. STXBP1 and STX1B both encode components of the SNARE complex, a complicated protein complex that mediates the fusion of the plasma membrane of the presynapse and the synaptic vesicle to allow for neurotransmitter release. DNM1, encoding the dynamin-1 protein, plays an essential role in recycling synaptic vesicles back into the presynapse after neurotransmitter release. A new study by Myers and collaborators has identified several patients with de novo variants in PPP3CA, which encodes another protein involved in synaptic vesicle recycling, further highlighting the importance of synaptic transmission in the etiology of severe neurodevelopmental disorders. In the interest of full disclosure, I am also a co-author on this study.
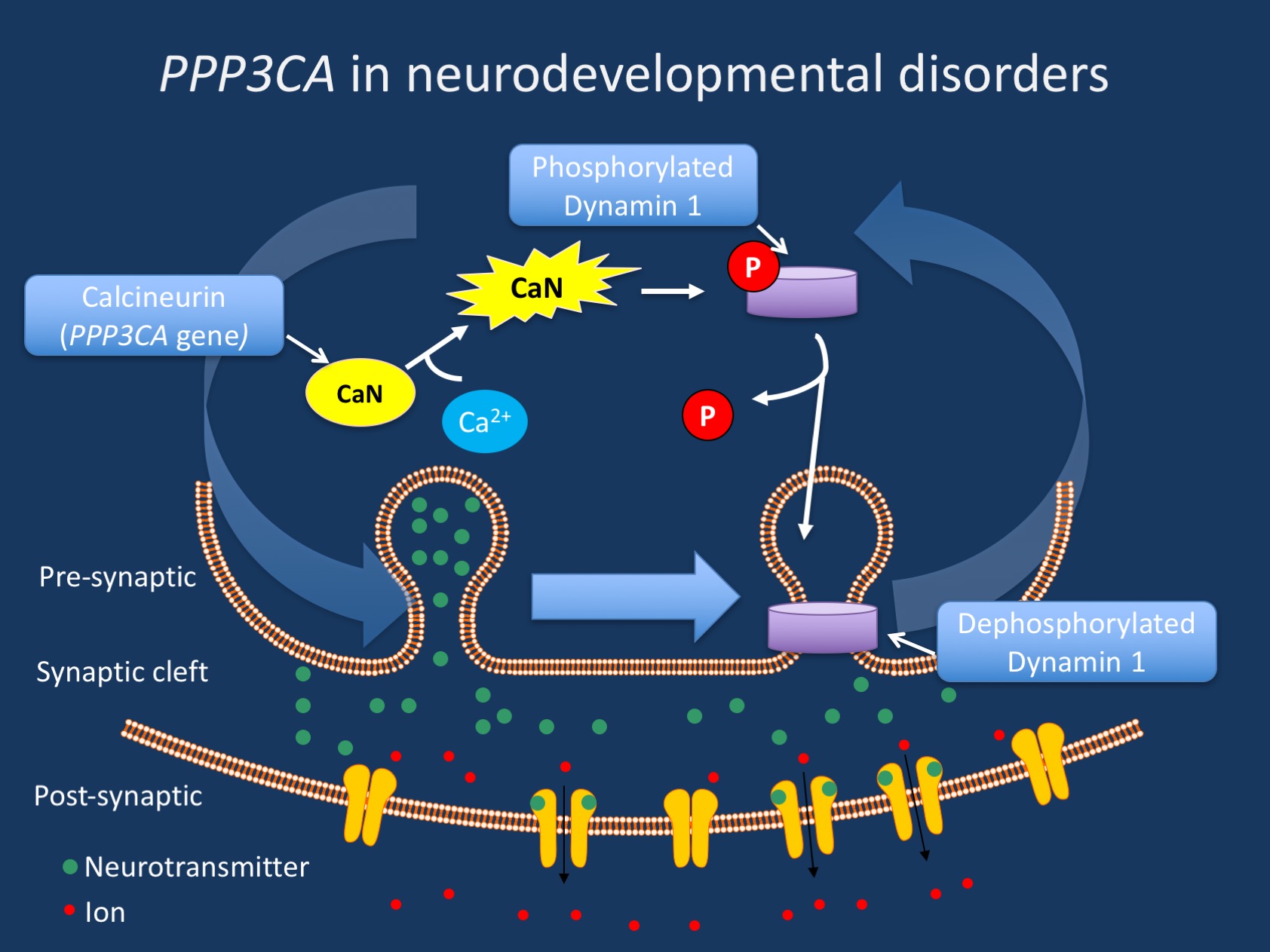
Figure. PPP3CA encodes the catalytic A subunit of the enzyme calcineurin shown above in yellow. Calcineurin is a calcium dependent serine-threonine phosphatase that modulates dynamin-1 mediated synaptic vesicle recycling. After nerve depolarization, calcineurin responds to an increase in calcium ions in the presynapse and removes the phosphate group (red) from inactive dynamin-1 (purple). The now dephosphorylated dynamin-1 is active and is able to participate in synaptic vesicle fission.
Calcineurin and dynamin-1 interaction. PPP3CA encodes the catalytic A subunit of calcineurin, a calcium- and calmodulin-dependent serine/threonine phosphatase. Calcineurin is known to play an important role in T-cell activation. Many readers may recognize calcineurin because it is the target of calcineurin inhibiting drugs, including cyclosporin. However, calcineurin plays another critical role in the brain and is responsible for the calcium-dependent dephosphorylation of dynamin-1 after nerve depolarization. As we have discussed previously on this blog, DNM1 is a critical protein in vesicle fission. Dynamin-1 proteins form a functional clamp around the base of the synaptic vesicle, fused to the plasma membrane, after neurotransmitter release. This dynamin clamp then twists tighter and tighter until the vesicle pops off and is recycled back into the presynapse. Calcineurin plays a crucial role in this process; in a phosphorylated state, dynamin-1 remains inactive in the cytoplasm. However, after nerve depolarization, calcineurin dephosphorylates dynamin-1, kicking it into its active state.
The PPP3CA phenotype. What is the clinical picture of these patients, and does it resemble DNM1 encephalopathy? Myers and collaborators identify six unrelated patients with de novo PPP3CA variants. Five of these individuals came from a cohort of over 4,700 individuals with neurodevelopmental disorders, which is significantly more de novo variation in PPP3CA than would be expected by chance. All individuals have severe neurodevelopmental disorders; five out of six have epilepsy. Although the clinical picture is in some ways similar to DNM1 encephalopathy – all affected individuals have severe to profound intellectual disability and 3/6 presented with a developmental and epileptic encephalopathy picture – the phenotype of PPP3CA is less homogeneous than what we have seen so far in DNM1, where most patients described so far have infantile spasms and/or Lennox-Gastaut syndrome. The epilepsy phenotype associated with pathogenic variants in PPP3CA, at least based on these five patients, was more variable with no consistent pattern emerging in terms of age of onset, frequency, or type of seizures observed.
Functional insights. The functional mechanism of the identified PPP3CA variants remains to be elucidated. However, it is notable that a recurrent variant (Gln282Lys) was reported in two individuals. The active site of PPP3CA is coordinated by three histidine amino acid residues (His92, His199, His281), one asparagine (Asn150), and two aspartic acids (Asp90, Asp118). Four of the six reported individuals in this study have de novo variants affecting these or adjacent amino acid residues, suggesting that these variants may interfere with the enzymatic properties of the protein.
What you need to know. Myers and collaborators have identified six unrelated individuals with de novo missense variants in PPP3CA, which encodes the catalytic A subunit of calcineurin. Calcineurin is responsible for calcium-dependent dephosphorylation of dynamin-1 and therefore plays an important role in synaptic vesicle recycling. This study implicates another synaptic transmission-related gene in the etiology of severe neurodevelopmental disorders, further emphasizing the biological importance of this pathway in neurodevelopment.