
Power outage. This week’s publications of the week were conceptualized in complete darkness. A thunderstorm had hit the Philadelphia area on Tuesday, leading to widespread power outages in the region. I found myself in the strange position of being without power for a night, but with full strength cell phone reception and a completely charged laptop battery. Here is our post on the most relevant publications of the last few weeks, written in the calm of a dark night where the only sound around was the howling of our neighbor’s backup generator.
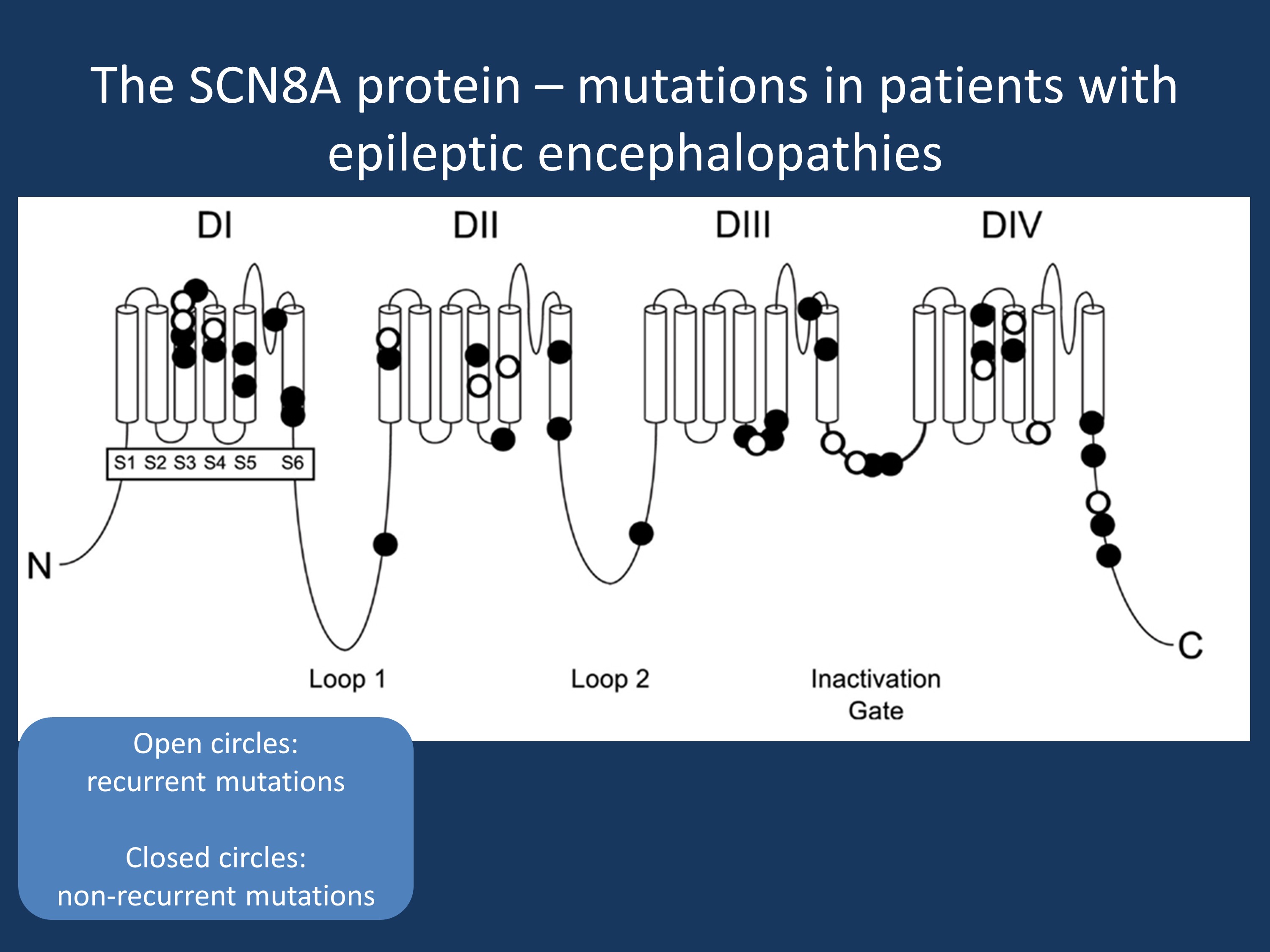
The SCN8A ion channel. The SCN8A protein (Nav1.6) contains four homologous domains with six transmembrane segments each (S1 to S6). Loop 3, linking DIII and DIV (labelled as “inactivation gate”), is highly conserved and functions as the inactivation gate. Loops 1 and 2 are less conserved than the transmembrane and linker domains from an evolutionary perspective. Closed symbols, non-recurrent SCN8A mutations; open symbols, recurrent SCN8A mutations. Adapted from Wagnon and Meisler from their open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). In accordance with the licensing for this article by Frontiers in Neurology, we have credited the authors and cited the journal in accordance with accepted academic practice.
Recurrent mutations in SCN8A. In a recent publication in Frontiers of Neurology, Wagnon and Meisler review the current knowledge about recurrent and non-recurrent mutation in SCN8A in epileptic encephalopathy. They find that roughly one third of patients with SCN8A encephalopathy have recurrent de novo mutations, which seem to cluster at CpG islands. The recurrent mutations are p.Asn215Asp, p.Ala890Thr, p.Ile132Val, p.Arg1617Gln, p.Ala1650Thr, and p.Arg1872Trp (with other amino acid substitutions at the same site such as p.Arg1872Gln, p.Arg1872Leu). The p.Arg1872Trp is the most common mutation and has been reported nine times in the literature. In particular, the phenotypic variability of the p.Arg1617Gln mutation seems to be quite high. Seizures in patients with these mutations had variable onset between 3 months and 12 months and treatment response to sodium channel blockers was also variable, ranging from treatment-responsive to refractory.
Comment: SCN8A epileptic encephalopathy is a disease of recurrent de novo mutations. However, even in patients with identical mutations, there is some phenotypic variability. The review by Wagnon and Meisler highlights that CpG islands are an important genetic feature predisposing to recurrent de novo mutations.
SYN1 and bathing epilepsy. Mutations in the SYN1 gene were identified as a cause of X-linked intellectual disability and epilepsy. In a recent publication in Epilepsia, Nguyen and collaborators further detail the phenotypic features of patients carrying SYN1 mutations. In addition to complex partial seizures, they find that seven out of ten family members with epilepsy had reflex seizures triggered by bathing or showering. Bathing seizures are a very rare seizures phenotype that is sometimes associated with Dravet Syndrome. However, in the current French-Canadian family, this seizure type was the only other seizure type in addition to complex partial seizures.
Comment: The connection of SYN1 mutations and a very rare seizure type is very interesting. It will be very interesting to see whether mutations in this gene can also be found in other unsolved cases with reflex epilepsy.
ZDHHC9 and rolandic epilepsy. ZDHHC9 is one of the many genes for X-linked intellectual disability. In a recent publication in the Annals of Clinical and Translational Neurology, Baker and collaborators review the phenotypic features of patients with ZDHHC9 mutations. They find that seven out of nine patients with mutations in this gene had epilepsy. Furthermore, the characteristics of the patients’ seizures were compatible with Rolandic Epilepsy (RE). The patients also had neurocognitive deficits that were most prominent in the regions usually affected by RE including impaired oromotor control.
Comment: Another way of looking at ZDHHC9 is to conceptualize this gene as a gene for rolandic epilepsy with intellectual disability. Further research will tell us whether this gene also carries susceptibility to rolandic epilepsy without intellectual disability and whether it can also be identified in patients with Electrical Status Epilepticus during Slow-Wave Sleep (ESES).
SCNM1 and reflex epilepsy. In a recent publication in Seizure, Kasteleijn-Nolte Trenite and collaborators present the clinical and genetic analysis of a family with two rare reflex epilepsies. All nine affected family members had a strong photoparoxysmal response, and four affected individuals had a combination of speaking-induced jaw-jerks and limb jerks evoked by flickering sunlight, often occurring during later in life in their 40s and 50s. Genetic analysis suggested a shared variant in SCNM1, a gene coding for a protein involved in splicing of sodium channels. This rare variant has a low population frequency in the European population and was found in 32/66,000 individuals in the ExAC browser.
Comment: The combination of the reflex seizures in this family is unique and may present a highly exaggerated photoparoxysmal response. It will be very interesting to see whether the genetic finding can be replicated in other families and to assess the phenotype of individuals in the population carrying this rare variant.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
