
In depth. Last week, we briefly mentioned the publication by Reinthalter and collaborators on 16p11.2 duplications in Benign Rolandic Epilepsy. At first glance, you might think that Eva’s publication may just be another description of a microdeletion in another type of epilepsy. However, nothing could be further from the truth. It’s a game changer. Here are three reasons why.
A brief summary. 16p11.2 microduplications arise due to the peculiar genomic architecture of human chromosome 16, which is covered with segmental duplications. These segmental duplications provide default breakpoints for small structural rearrangements, which lead to microdeletions and microduplications. In a recent publication in Human Molecular Genetics, Reinthaler and collaborators identify 16p11.2 microduplications as a major risk factor for Benign Rolandic Epilepsy, one of the most common epilepsies of childhood. These duplication are found in ~1% of patients and are virtually absent in controls. These are the three ways the publication by Reinthaler and collaborators changed our way of thinking.
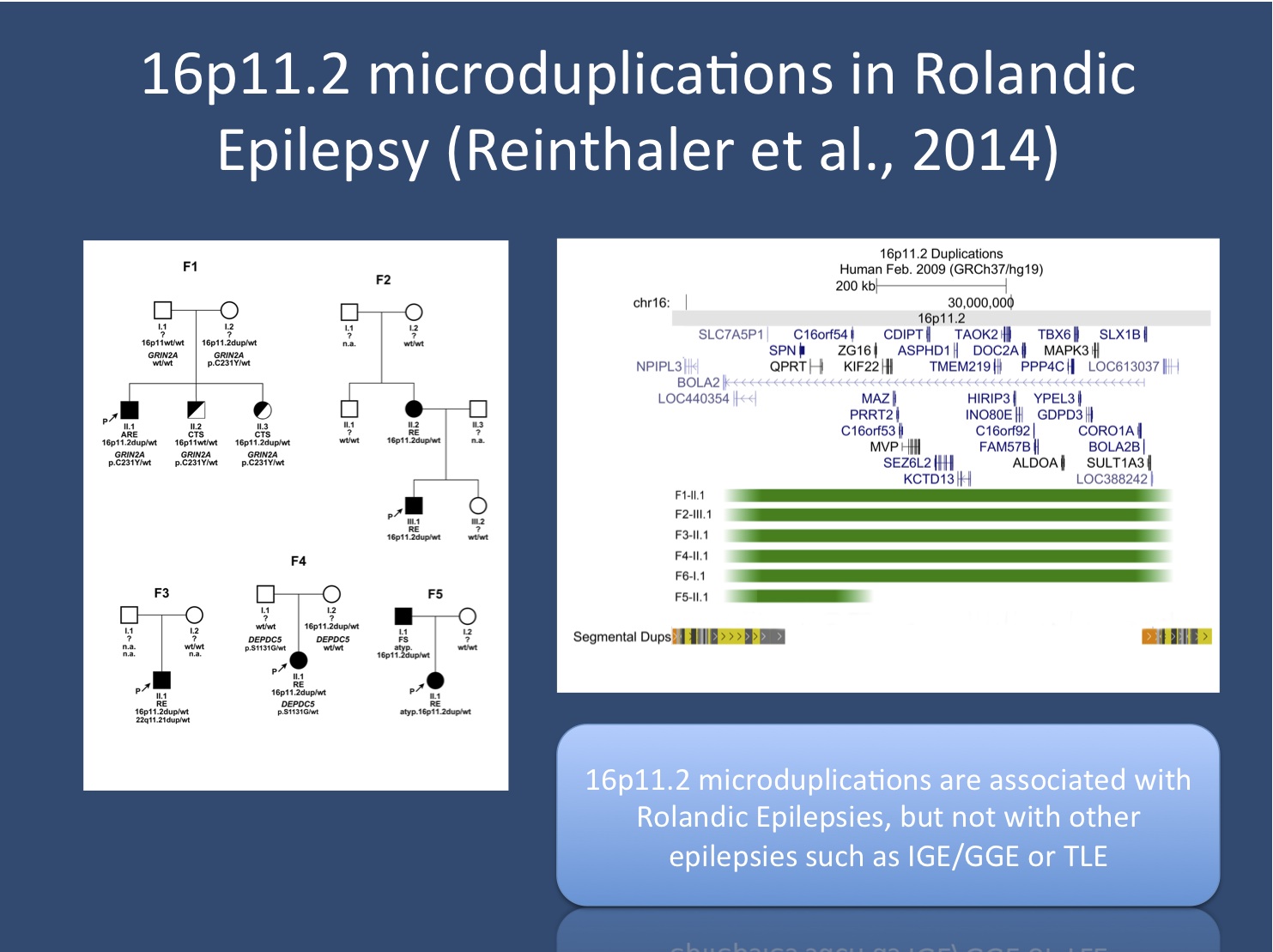
Pedigrees of families with 16p11.2 microduplications and rolandic epilepsy. As with other microdeletions and microduplication, most variants are inherited, often from non-affected parents. This is a pattern expected in rare genetic risk factors and the support for their role as risk factors comes from association studies, not family studies. On the right, the microduplications identified in patients are shown including the possible candidate genes within the 16p11.2 microdups (image provided by author).
1 – Rolandic Epilepsy, finally. It’s good to see a genetic risk factor for Benign Rolandic Epilepsy, as research into the genetics of Rolandic Epilepsy has never been easy. Or to put it differently: it has been as complicated as things can get. While there are intuitive examples for a genetic contribution in other types of non-lesional epilepsy such as identical twins with Childhood Absence Epilepsy reported by William Lennox, there is no such thing with Rolandic Epilepsy. Basically, there is no single identical twin pair reported with both twins affected by Rolandic Epilepsy – not the best place to start the hunt for epilepsy genes. It became eventually clear that the EEG phenotype of Rolandic Epilepsy, centrotemporal spikes, would be the more reliable parameter for genetic studies. However, despite promising initial studies, this field of research never moved beyond the stage of linkage studies. While it was undeniable that there must be some genetic contribution, Rolandic Epilepsy remained the big mystery. That’s why the study by Reinthaler and collaborators is a first crucial step.
2 – A microduplication, who would have thought? I would not have been surprised if the first CNV associated with BRE had been a microdeletion, maybe even the 16p13.11 or 15q13.3 microdeletion. However, it turned out to be the 16p11.2 microduplication. In contrast to the corresponding microdeletions, the role of microduplications is often more difficult to track down. The risk conferred by microduplicaitons is usually smaller and the phenotypes less clear. Things seem to be different for the 16p11.2 microduplication, though. The strong connection with BRE is surprising, as the phenotype is not associated with the corresponding microdeletion and is only found in a small subset of duplication carriers. A constellation like this would have been impossible to track down only a few years ago. Now, with increasing sample sizes, supporting these findings with sufficient statistical rigor is manageable. There are basically tens of thousands of individuals genotyped – it’s just important to put all the data together. This is what Reinthaler and collaborators did.
3 – “Benign” epilepsies still on the agenda. Over the last four years, there was a major shift towards gene identification in severe, early-onset epilepsies, which has led to the discovery of de novo mutations in multiple causative genes in these disorders. Prior to this recent surge in gene discovery, the more “benign” and common epilepsies were considered to be the ideal model system for epilepsy genetics. The general way of thinking was thatby investigating the genetics of common epilepsies, we will discover new mechanisms that would then be applicable to the rare epilepsies, as well. However, this strategy didn’t seem to work. While we have witnessed a virtual explosion of genes for rare epileptic encephalopathies, we are still struggling to get a hold of genetic risk factors for common epilepsies. In some areas such as the genetics of common Febrile Seizures, joint efforts for gene discovery have stopped altogether. The publication of Reinthaler and collaborators is a good reminder that gene discovery in common, “benign” epilepsies is possible and worthwhile and may lead to meaningful results. It’s a good motivator to continue on our quest to also understand the genetics of the more “benign” epilepsies. By the way, I use “benign” in quotation marks – any type of epilepsy is far from benign.
Addendum: What about GRIN2A and Rolandic epilepsy? You might have realized that we have labelled the 16p11.2 microdup the first true genetic risk factor for Rolandic Epilepsy and have not mentioned GRIN2A. Mutations in GRIN2A are associated with epilepsy-aphasia syndromes, which represent the severe end of Rolandic epilepsies. However, these conditions are distinct from the more common Rolandic epilepsies. GRIN2A variants can be found in patients with common Rolandic epilepsies, but demonstrating that this is an enrichment compared to controls would require large sample sizes. It is not clear at the moment whether some GRIN2A variants would be considered strong enough in Rolandic epilepsy to be called causative. I also didn’t mention DEPDC5, a gene for familial focal epilepsies. Variants in DEPDC5 were recently identified in families with BRE. However, it is extremely difficult to pinpoint the causative nature of such variants in small families with incomplete penetrance.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany

